
![]() # Gene Set Clustering based on Functional annotation
# Gene Set Clustering based on Functional annotation
======================================================================
GeneSCF serves as command line tool for clustering the list of genes given by the users based on functional annotation (Gene Ontology, KEGG, REACTOME and NCG 4.0). It requires gene list in the form of Entrez Gene IDs or Official gene symbols as a input. GeneSCF supports multiple organisms from V1.1. Examples to download database as simple text file using GeneSCF "prepare_database" module, 1) E.coli 2) Sheep , 3) General usage
The advantage of using GeneSCF over other enrichment tools is that, it performs enrichment analysis in real-time (v1.1 and above) by accessing source databases. With command-line versions of tools, as you know you can run multiple gene list simultaneously.
Please follow GeneSCF news section on Biostars or  to get latest updates on GeneSCF .
to get latest updates on GeneSCF .
======================================================================
Home page: https://github.com/genescf
Requirement:
GeneSCF only works on Linux system, it has been successfully tested on Ubuntu, Mint,Cent OS and Windows 10 bash (version 1607). Other distributions of Linux might work as well.
Documentation: https://github.com/genescf
Cite using: Subhash S and Kanduri C. GeneSCF: a real-time based functional enrichment tool with support for multiple organisms. BMC Bioinformatics 2016, 17:365, http://www.biomedcentral.com/1471-2105/17/365
Report issues in Biostars or GitHub Project page.
Discussions on Biostars
- GeneSCF gives out more pathways for genes compared to DAVID
- More .... https://www.biostars.org/local/search/page/?q=genescf
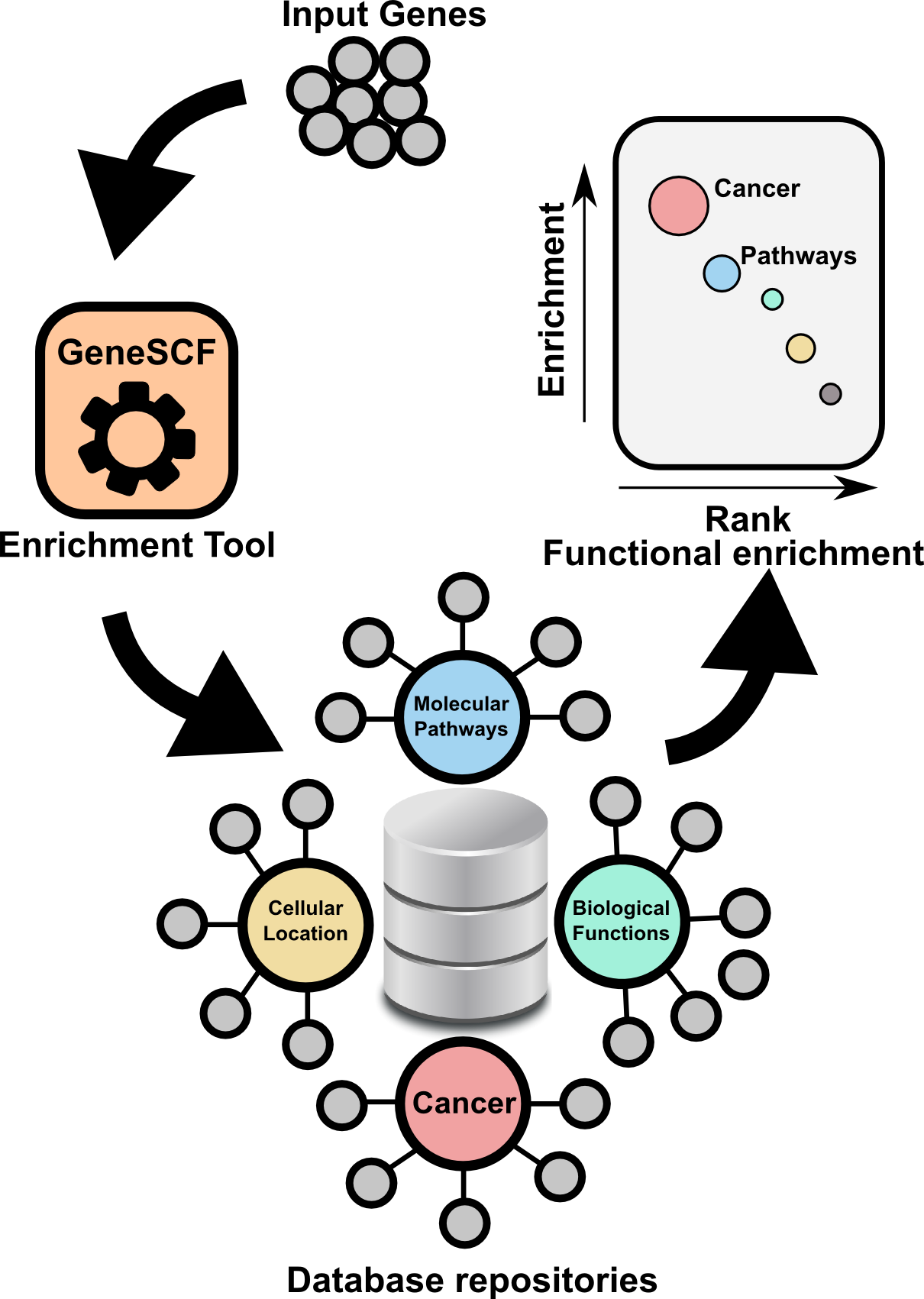
======================================================================
Advantages
- Real-time analysis, do not have to depend on enrichment tools to get updated.
- Easy for computational biologists to integrate this simple tool with their NGS pipeline.
- GeneSCF supports more organisms.
- Enrichment analysis for Multiple gene list in single run.
- Enrichment analysis for Multiple gene list using Multiple source database (GO,KEGG, REACTOME and NCG) in single run.
- Download complete GO terms/Pathways/Functions with associated genes as simple table format in a plain text file (Check "Two step process" below in "GeneSCF USAGE" section).
======================================================================
Get organism codes for GeneSCF run
KEGG: Second column from the following link. For human 'hsa' and Mus Musculus 'mmu'.
http://rest.kegg.jp/list/organism
Gene Ontology: Use "id" from the following link. Example for human "goa_human" and "mgi" for Mus Musculus.
http://www.geneontology.org/gene-associations/go_annotation_metadata.all.json
======================================================================
Comparison (updated on Tue Jul 26 16:01:08 CEST 2016)
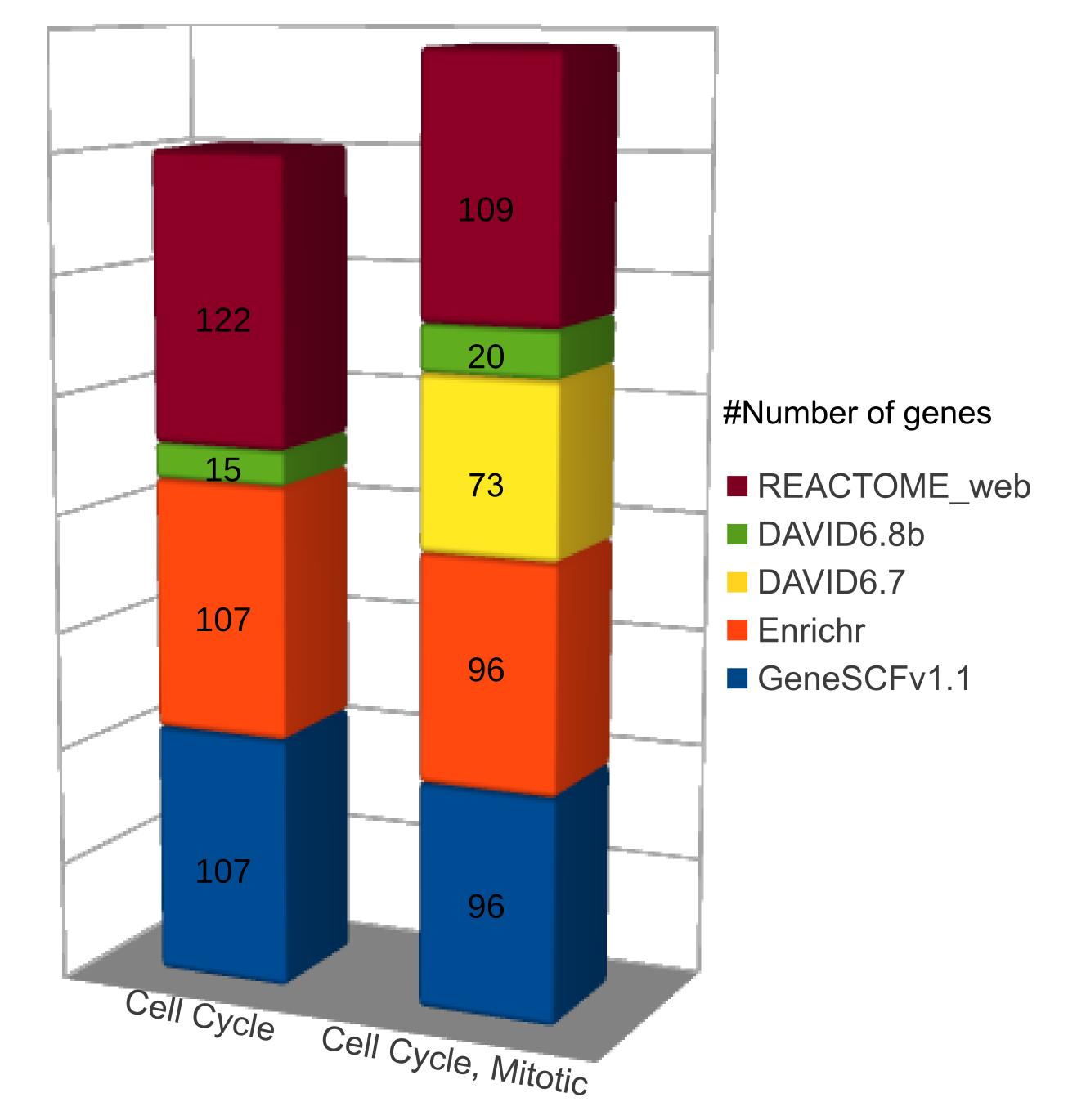
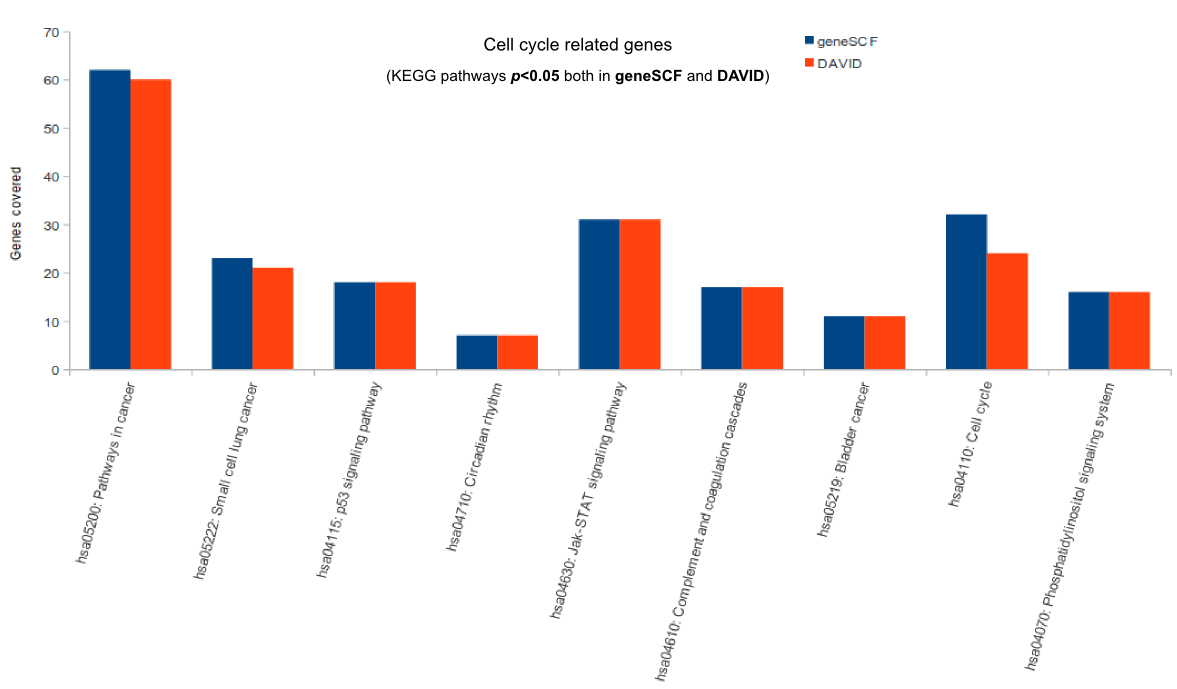
For more comparisons please check GeneSCF article (Fig. 6).
======================================================================
GeneSCF USAGE
Example (using GeneSCF v1.1 and above)
I will use example for Mus musculus assuming you got Entrez geneids,
Single step process,
Gene Ontology - Biological Process (Downloading current available database for Mus Musculus from Gene Ontology + enrichment analysis)
./geneSCF \
-m=update \
-i=INPUTgene.list \
-t=gid \
-db=GO_BP \
-o=/ExistingOUTPUTfolder/ \
-org=mgi \
--plot=yes \
--background=15000
The above command downloads complete GO db as simple text file in following location, geneSCF-tool/class/lib/db/mgi/ and also do enrichment analysis parallel. The results for enrichment analysis can be found in folder ExistingOUTPUTfolder.
No need for running update mode for consecutive runs since GO database for Mus musculus got updated when you use update mode on first run.
Gene Ontology - Cellular Component
./geneSCF \
-m=normal \
-i=INPUTgene.list \
-t=gid \
-db=GO_CC \
-o=/ExistingOUTPUTfolder/ \
-org=mgi \
--plot=yes \
--background=15000
Gene Ontology - Molecular Function
./geneSCF \
-m=normal \
-i=INPUTgene.list \
-t=gid \
-db=GO_MF \
-o=/ExistingOUTPUTfolder/ \
-org=mgi \
--plot=yes \
--background=15000
Gene Ontology - Complete (BP+CC+MF)
./geneSCF \
-m=normal \
-i=INPUTgene.list \
-t=gid \
-db=GO_all \
-o=/ExistingOUTPUTfolder/ \
-org=mgi \
--plot=yes \
--background=15000
Two step process,
Downloading current available database for Mus Musculus from Gene Ontology
./prepare_database -db=GO_all -org=mgi
The above command downloads complete GO db as simple text file in following location, geneSCF-tool/class/lib/db/mgi/.
Gene Ontology - Biological Process
./geneSCF \
-m=normal \
-i=INPUTgene.list \
-t=gid \
-db=GO_BP \
-o=/ExistingOUTPUTfolder/ \
-org=mgi \
--plot=yes \
--background=15000
Gene Ontology - Cellular Component
./geneSCF \
-m=normal \
-i=INPUTgene.list \
-t=gid \
-db=GO_CC \
-o=/ExistingOUTPUTfolder/ \
-org=mgi \
--plot=yes \
--background=15000
Gene Ontology - Molecular Function
./geneSCF \
-m=normal \
-i=INPUTgene.list \
-t=gid \
-db=GO_MF \
-o=/ExistingOUTPUTfolder/ \
-org=mgi \
--plot=yes \
--background=15000
Gene Ontology - Complete (BP+CC+MF)
./geneSCF \
-m=normal \
-i=INPUTgene.list \
-t=gid \
-db=GO_all \
-o=/ExistingOUTPUTfolder/ \
-org=mgi \
--plot=yes \
--background=15000
The results for enrichment analysis can be found in folder ExistingOUTPUTfolder.
The above mentioned parameters should be changed according to your data (following can be altered),
-t=sym (for Gene Symbol as input list)
-t=gid (for Entrez Geneid as input list)
--background=#NUM (Use the total number of background genes from your dataset, example you can use total number of protein coding genes with detectable expression level irrespective of their significance or if it is transcriptome/Genome wide study you can use total number of annotated protein coding genes as background)
More information please refer documentation, https://github.com/genescf
======================================================================
Instructions for running batch analysis (Supported above GeneSCF v1.1 patch release 2 - GeneSCF v1.1-p2)
Edit script
./geneSCF-master-source-v1.1-p2/geneSCF_batchfor your input files (files_path) and output path (output_path).- files_path="/FOLDER/WHERE/GENE_LISTS/STORED"
- output_path="/FOLDER/PATH/FOR/OUTPUT"
Edit file
./geneSCF-master-source-v1.1-p2/db_batch_config.txtto configure your parameters for batch run.- Execute [genescf_path]/geneSCF-master-source-v1.1-p2/geneSCF_batch.
Note:
- Recommended to keep all input files in same folder.
- Inside specified output folder path GeneSCF will automatically create individual sub-folders for each gene list.
======================================================================
This works for plants?
Now GeneSCF v1.1 supports multiple species/organisms. Check out.
is it convinient for you to server a list of supported species
Hi
KEGG: http://rest.kegg.jp/list/organism
Gene Ontology: http://www.geneontology.org/gene-associations/go_annotation_metadata.all.json
Reactome & NCG: Human only
You can also find this information on GeneSCF documentation under heading '3. Preparing database'. Also the lists are provided with GeneSCF tool download in directory 'org_codes_help'. To get updated list always follow the links from the documentation.
Hello, I have several questions here.
First, have you compared your tool to DAVID? I know that DAVID uses a bit outdated GO annotation, but still are we about to get more precise annotation with geneSCF?
Second, does your software allow integrating of annotation from different sources. I mean there are certain pieces of information that are in different annotation sets that are missing and therefore annotation sets could complement each other.
Third, any details, besides source code, how the clustering is performed. I've seen the term EASE in your wiki, which I believe is a clustering algorithm implemented by DAVID itself. How is this incorporated into your framework?
Hi mikhail.shugay
I hope answered for all your question. Please let me know if you have more questions.
Hi i'm getting this error when I tried to use your program
This is the command that I used
my input
./geneSCF --infile=test_entrezID.txt --gtype=gid --outpath=~/EC_gene/ --database=GO_all
Please rewrite the command as above.
Also instead of 'gtype' you wrote 'gytpe'
I also getting same error when I run following command using same above input file test_entrezID.txt :
also tried with -m=update
Illegal division by zero at /geneSCF-master-source-v1.1/class/lib/List/Vectorize/lib/List.pl line 599, <in2> chunk 1.
Is it possible to provide few lines of your input gene list ?
Check whether you have provided the gene lists in proper format, I am afraid there might be problem with your input. Examples for preferred formats are in this link
https://github.com/santhilalsubhash/geneSCF/tree/master/geneSCF-master-v1.0/test
Entrez GeneID format: sample_gene_list_id ( format supported by --gtype=gid )
Gene symbol format: sample_gene_list_sym ( format supported by --gtype=sym )
I used same input as in above example: test_entrezID.txt
test_entrezID.txt
Please try to use full path or if it is in the current directory use,
./geneSCF -m=normal --infile=./test_entrezID.txt --gtype=gid --outpath=./test/output/ --database=GO_all --plot=yes -bg=20000 -org=goa_human
Let me know if it is working.
and this is command & same error
Illegal division by zero at /geneSCF-master-source-v1.1/class/lib/List/Vectorize/lib/List.pl line 599, <in2> chunk 1.
Can you please try the tutorial on test dataset from GeneSCF v1.1 by redownloading the tool?
http://genescf.kandurilab.org/downloads.php
http://genescf.kandurilab.org/documentation.php
I also want to point out that GeneSCF only works on LINUX system, it has been successfully tested on UBUNTU, MINT and CentOS. Other distributions of Linux might work as well.
Thanks for your prompt response, actually I was trying on mac os terminal.
We are sorry for not being clear, soon we will update the information in documentation and in the website as well. The future version of GeneSCF can be made available for OSX operating environment (current versions only works on Linux environment).
Dear EagleEye, Hi
Is it possible to use your tool in the case of de novo transcriptome assembly Clustering of Functional annotation ?
I have a fish de novo assembly (transcripts) and I don't know how to use geneSCF for my data (I usually use Blast2GO for annotation and then WEGO for visualization).
~ Thanks
GeneSCF works only with simple gene list from known organism or species covered by KEGG and GeneOntology (GO). If you have gene list predicted from your analysis, you can use one of the organism from the below links close to your organism (fish) as model to predict function or perform enrichment analysis.
For,
Organisms/species covered by KEGG
Organisms/species covered by GO
Also the links are provided in the post under heading 'Get organism codes for GeneSCF run'.
Dear EagleEye,
I guess the most annotated organism close to my species in zebrafish.
I can blast my transcriptome.fasta against ENSEMBL zebrafish genes ( or proteins) database and collect the related gene (in zebrafish).
1- can I use the data I have described in GeneSCF ?
2- imagine that I have this Gene list, what is the simple script for running GeneSCF for my data ?
Thank you
YES. You can use your list of collected related genes from zebra fish (Danio rerio) in GeneSCF.
GeneSCF commandline for zebra fish (Danio rerio), organism code is 'dre' from KEGG.
./geneSCF -m=update -i=INPUTgene.list -t=gid -db=KEGG -o=/ExistingOUTPUTfolder/ -org=dre --plot=yes --background=15000
'INPUTgene.list' is a file with list of your genes.
'ExistingOUTPUTfolder' already existing folder where your output to be stored.
For background instead of 15,000 use th total number of genes found by transcriptome assembly.
'-t=gid' should be changed according to your input type.
Also check system requirements for running GeneSCF.
How much does it take to run? Is there any benchmarking? It took 5 minutes to analyze 2000 genes. How does it scale?
Sorry for my late response (On vacation :-))
Simulations performed on March 2016 using GeneSCF v1.1
Hi there, I was trying to run genescf. 1st I tried with my raw csv file for multiple organisms. But getting errors. I have changed my csv format to text , each line one gene name and ran it for single organism. Still getting the error. anybody can help me? Is there any way to run it for multiple organisms?
Best Regards Zillur
error:
cat: /home/zillur/Desktop/zillur/phd/orthofinder/genescf/geneSCF-master-source-v1.1-p2/class/lib/db/pfa/gene_association.pfa: No such file or directory cat: /home/zillur/Desktop/zillur/phd/orthofinder/genescf/geneSCF-master-source-v1.1-p2/class/lib/db/pfa/gene_association.pfa: No such file or directory Updating gene information... Do not panic. The processing is going on..
Tue Feb 28 02:21:18 AST 2017 finished processing
Please provide complete path to input file (single column file containing one geneid/symbol per line) and output path/folder (output path should end with "/").
Always check organism codes before running geneSCF. The code for your organism is "GeneDB_Pfalciparum".
Note: For organism codes please check the link provided in the documentation or org_code_help folder and link provided in the files for getting updated Organism codes.
And refer the detailed answers provided for your question on other thread.
Thank you very much. It works perfectly, exactly what I wanted. I have other organism's gene name in my list (Plasmodium, Cryptosporadium, Toxoplasma, Babesia etc). How can I map my gene list for other organisms? In the organism codes we have only GeneDB_Pfalciparum. Is there any way to map for other organisms also? Thanks again for help.
Best Regards Zillur
Hello!
Thank you very much for making geneSCF, it works really well!
I am currently trying to prepare the "goa_uniprot_all" database, which is quite large. I tried a few times before using a cluster, but the job could not finish for a long time. Do you have an estimated time for how long it would take the prepare this database?
Hi amy.bashir,
I am happy to know that you found this tool useful.
Since performance of GeneSCF 'prepare_database' or 'geneSCF update mode' modules are highly dependent on the connection with the source database and user internet bandwidth, it is hard for me to tell.
The 'goa_uniprot_all' is quite heavy load, I would recommend you to use as background process (example, 'nohup') if you have access to any cluster/sever.
Thank you very much for your quick reply! I will try that!
Hello!
I started the prepare_database job for goa_uniprot_all about 9 hours ago when I saw your answer. This is the command I used:
The database seems to have been downloaded without problem, but it's been stuck without any change after about 2 hours. I have attached a screenshot of the goa_uniprot_all folder as it is now (screenshot). I see the same thing when I tried to prepare this database before too. Can you please let me know what could be the issue?
Hi,
Whenever you use '
nohup', you should use ';' or '&' in the end. So that nohup will know it should exit or go to next command after it finishes the current program.Example,
In your case second one will work. I will better recommend you to run again with '&' before assuming and exit the command.
Great, thank you very much, I am trying that now!
Hello,
As you recommended, I tried this command to prepare the goa_uniprot_all database:
I also tried to use "update" mode along with a gene list, but in both cases, I see that the database files get stuck in the state as shown in the screenshot I included in the previous post. What should the properly prepared database for goa_uniprot_all look like?
Thank you very much for answering all my questions!
COMMAND-: ./prepare_database -db=GO -org=goa_human ERROR-: rm: cannot remove '/home/ibab/software/geneSCF-master-source-v1.1-p2/class/lib/db/gene_info_limit.gz': No such file or directory
Hello everyone!!!! I read the above conversation still i m confused as to why i m getting this error. I tried downloading it twice still the error persist. On carrying on further I m getting the following error.
ERROR -: Illegal division by zero at genescf/geneSCF-master-source-v1.1-p2/class/lib/List/Vectorize/lib/List.pl line 599, <in2> chunk 1.
Please guide me.
Hi,
COMMAND-: ./prepare_database -db=GO -org=goa_human ERROR-: rm: cannot remove '/home/ibab/software/geneSCF-master-source-v1.1-p2/class/lib/db/gene_info_limit.gz': No such file or directory
Answer: Do not worry about this error. When it is not found, GeneSCF can download the database.
ERROR -: Illegal division by zero at genescf/geneSCF-master-source-v1.1-p2/class/lib/List/Vectorize/lib/List.pl line 599, <in2> chunk 1.
Answer: Please provide your sample input gene list. Also let me know your operating system.
OS -: Ubuntu 16.04
LIST -: NR_046289 NR_046290 NR_046292 NR_046293 NR_046294 NR_046295 NR_046297 NR_046298 NR_046299 NR_046308 NR_046310 NR_046311 NR_046312 NR_046313
Per line one gene-id is present
I have a list of 54000 gene-ids I want to annotate all the genes. Is there any other R package or command-line software that I can use?
HI,
I have a list of 54000 gene-ids I want to annotate all the genes. Is there any other R package or command-line software that I can use?
Answer: Make this as separate post/question on Biostars. You will get quick help.
Hi,
In GeneSCF you can only use Gene Symbols (Example) or Entrez GeneIDs (Example) as input. And the reason for the error you mentioned is because your input gene list does not match with the database consists of only Gene symbols and Entrez IDs (Check reported issue).
It is also clearly state in GeneSCF FAQs under 'How to handle error Illegal division by zero?'
Good luck with your analysis.
I have tried to do GO with genescf as I did for the same gene symbols list by DAVID, but results with genescf are very different, and no P-values is significant (based on EASE less than 0.05). need to mention that I have used mgi as -db. DO you have any idea about this conflict?
Which version of GeneSCF are you using ?
geneSCF-master-source-v1.1-p2
Hi, please check with p-values in both case or use the same criteria (p-value or FDR etc.,). I do not know how DAVID processes the annotation and handle the database. Also check how many number of genes each tool could match for individual terms. You can also verify GeneSCF results with some other tools other than DAVID to make sure the results are fine. I would say GeneSCF is more trustworthy and reliable as there are no in-house processing of database involved or it does not stores the database. It simply retrieves the database from the repositories and calculates simple fisher's exact test followed by p-value corrections by different methods.
Hello, I intend to use GeneSCF to perform enrichment analysis. I am trying to prepare the database (goa_chicken) prior to running the analysis but I get this message that reads
Extracting goa_chicken information... cat: /Users/fjodor/Desktop/geneSCF-master-source-v1.1-p2/class/lib/db/goa_chicken/gene_association.goa_chicken: No such file or directory. This is strange since I can see that the .gz version of this file exists in the db folder. I have also made sure to install all the necessary UNIX commands before running the software. The command I used for the enrichment analysis was/geneSCF -m=update -i=/input.txt -t=sym -o=/path_to_dir -db=GO_all -p=no -bg=20000 -org=goa_chickenand the command for preparing the database was/prepare_database -db=GO_all -org=goa_chicken.Any help with this issue would be appreciated. Thank you!
May I know which operating system you are using ?
MacOS Mojave 10.14.2
Requirement: GeneSCF only works on Linux system, it has been successfully tested on Ubuntu, Mint,Cent OS and Windows 10 bash (version 1607). Other distributions of Linux might work as well.