Entering edit mode

9.8 years ago
tomasfer
•
0
Hi,
I mapped reads (150 bp) to exon reference (in Geneious) and it is apparent that there are two different types of sequences, probably paralogues, each with high coverage
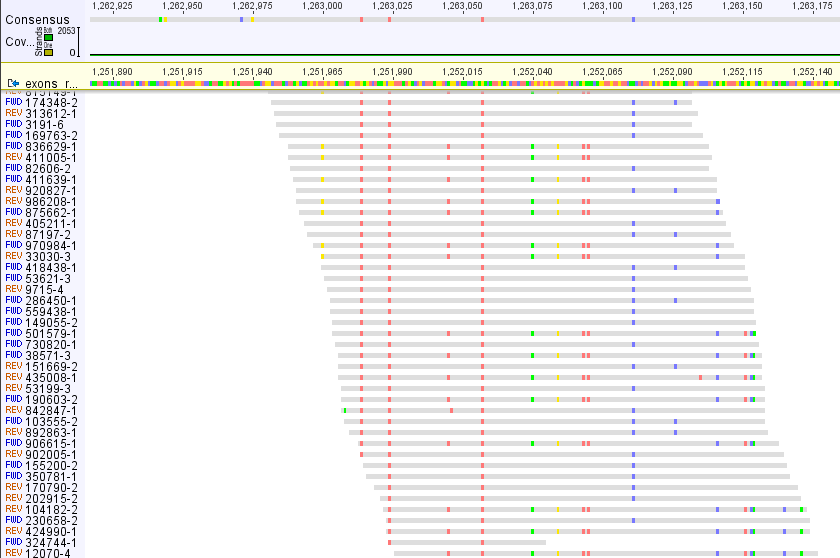
Making consensus does not make sense in this case, sequences will be used for phylogeny and paralogs can destroy true signal etc.
Is there any possibility how to extract these two sequences/paralogues from mapped reads, i.e. make an alignment (actually two alignments...) from these reads belonging only to particular variant.
I prefer some possibility that allows for streaming as I have several thousands of such loci (of course not all of them have two or more "paralogues").
Thanks for some suggestions or links.
Tomas

Did you align your sequences against the whole genome? Or only on this specific exon sequence?
My reads are from enriched library (ca. 2,500 exons, non-model species) so I mapped all reads to "pseudoreference" consisting from sequences used for enrichment separated by 400 Ns (to avoid reads to be mapped to multiple exons). What do you see on a picture is a part of a single exon.
Even with enriched libraries, you're highly advised to align to the entire genome. That alone may clear this up.
OK, but as I am working with non-model taxon, genome is not available :-(
Here, "genome" is whatever sort of reference you have, be it just contigs or something else.