I want to know what people who have used fastx or similar have done to define a poor quality read from RNA-seq. What do you call a bad base (I was thinking of calling one bad if it had a score of 20 or below from phred33) and how many bad bases do you allow per read? I wondered if >=1 bad Base per 10 would definite a bad read. So if I had a 100 bp read, 10 or more bases with a score or 20 or less would be rejected. Does this sound right? Too strict? To relaxed?
You're probably better off letting the aligner decide what a bad read is rather than throwing out some of your data because they don't meet some arbitrary criteria. If the aligner can unambiguously map the read, then does it matter if it has a few low quality bases in it? Base quality is also accounted for by downstream tools for which it's important (such as variant callers), so you normally don't have to worry about it yourself.
Often, however, it is a good idea to trim the reads from the end using a quality threshold, since base quality tends to drop off with the length of the read, particularly for illumina. Trimming will likely improve the mapping rate for aligners that map the entire read end-to-end, such as tophat, but not for those that can soft-clip the ends of reads to find the best match, such as STAR. I usually use cutadapt to do trimming and use a quality threshold of 25.
Thanks for the feedback. Generally I do let the aligner (Tophat) decide (I am glad to know others do as well). But these reads were particularly bad and I was worried that some were mismapping (I allow for a read to maps to multiple locations). I doubt it would be a major problem but thought people would be concerned if I left so many bad reads in there. Before mapping there is little to no bias of poor bases, they are throughout the whole read. After the mapping there is a slight bias towards the 3' end so trimming might help increase mapping percentage but I do not want that to come at the expense of incorrectly mapped reads. If you do not think that this is a problem, perhaps I will just leave it with the aligner doing the filtering.
According to the tophat manual, any reads which align to the reference with more than two mismatches are discarded. Unless you've changed this parameter, it seems to me that you would have to be very unlucky to have base error(s) that would lead to a read mapping almost perfectly to an incorrect location. Much more likely is that the read would just get discarded, which would have a similar effect as what you're trying to achieve by prefiltering. So I think the problem with low quality reads is not so much mismapping but rather data loss, but this is my just two cents and others with more experience might think differently. Have you run something like FastQC on your reads and found them to be flagged for low quality?
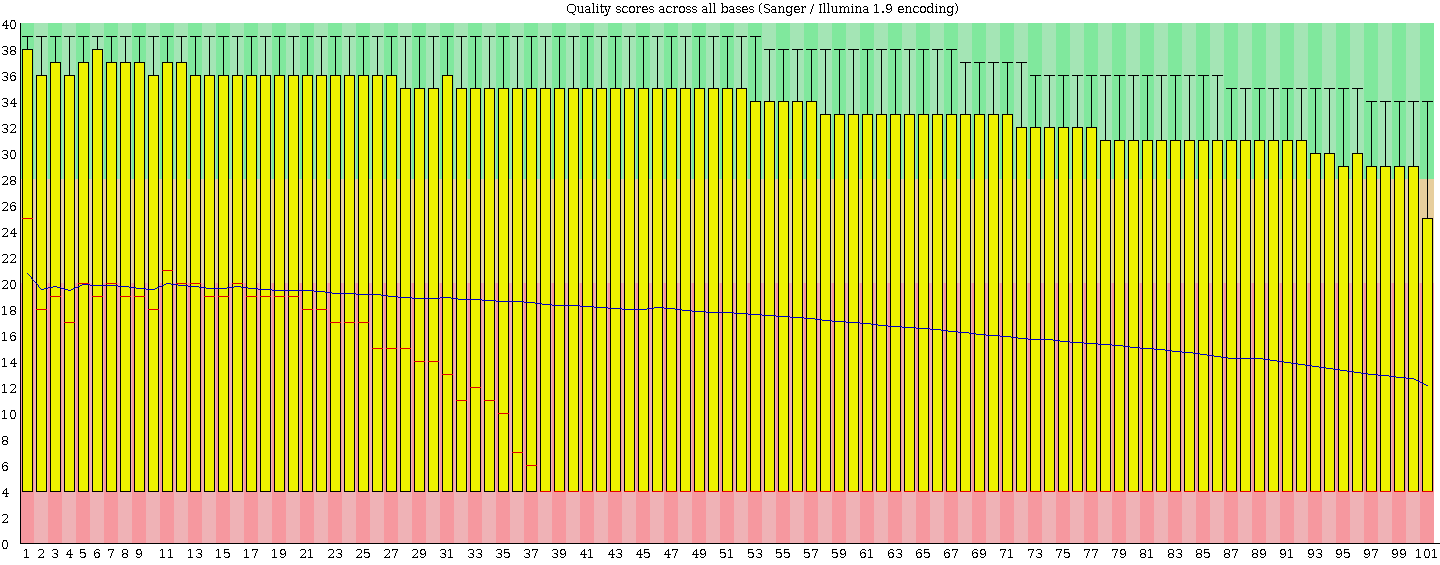
I stick to Tophat's two mismatches so I agree I would be very unlucky. Normally I take this approach but the high level of poor bases scared me and thought people (like my supervisor and other RNA boffins) would criticise not filtering. I will find out what they think tomorrow. I have ran FASTQC and this is how I noticed the poor reads (that and the low mapping %). the score pre-mapping is low across the whole reads. Below is an image from FASTQC on the unmapped reads (i.e. the whole fastq file).
I have a similar experience before. I tried different trimming threshold to get the maximum number of mapped reads. However, my conclusion is that a very low quality data could result in Garbage In and Garbage Out, no matter what methods we adopted. The below is a related paper for your reference.
de Sa, P.H., Veras, A.A., Carneiro, A.R., Pinheiro, K.C., Pinto, A.C., Soares, S.C., Schneider, M.P., Azevedo, V., Silva, A. & Ramos, R.T. (2015). The impact of quality filter for RNA-Seq. Gene.
ADD COMMENT
• link
updated 2.9 years ago by
Ram
45k
•
written 10.1 years ago by
Gary
▴
480
0
Entering edit mode
Thanks for the reference, I will certainly check it out. The results the original paper using this data published seemed to make sense from what we already know using different methods or from different organisms so I think there must be enough signal within all this noise to get something out of it.



Thanks for the feedback. Generally I do let the aligner (Tophat) decide (I am glad to know others do as well). But these reads were particularly bad and I was worried that some were mismapping (I allow for a read to maps to multiple locations). I doubt it would be a major problem but thought people would be concerned if I left so many bad reads in there. Before mapping there is little to no bias of poor bases, they are throughout the whole read. After the mapping there is a slight bias towards the 3' end so trimming might help increase mapping percentage but I do not want that to come at the expense of incorrectly mapped reads. If you do not think that this is a problem, perhaps I will just leave it with the aligner doing the filtering.
According to the tophat manual, any reads which align to the reference with more than two mismatches are discarded. Unless you've changed this parameter, it seems to me that you would have to be very unlucky to have base error(s) that would lead to a read mapping almost perfectly to an incorrect location. Much more likely is that the read would just get discarded, which would have a similar effect as what you're trying to achieve by prefiltering. So I think the problem with low quality reads is not so much mismapping but rather data loss, but this is my just two cents and others with more experience might think differently. Have you run something like FastQC on your reads and found them to be flagged for low quality?
I stick to Tophat's two mismatches so I agree I would be very unlucky. Normally I take this approach but the high level of poor bases scared me and thought people (like my supervisor and other RNA boffins) would criticise not filtering. I will find out what they think tomorrow. I have ran FASTQC and this is how I noticed the poor reads (that and the low mapping %). the score pre-mapping is low across the whole reads. Below is an image from FASTQC on the unmapped reads (i.e. the whole fastq file).