
In whole-genome high-throughput sequencing data, one expects a clear separation between high-frequency k-mers (signal) and low-frequency k-mers (noise arising from sequencing errors):
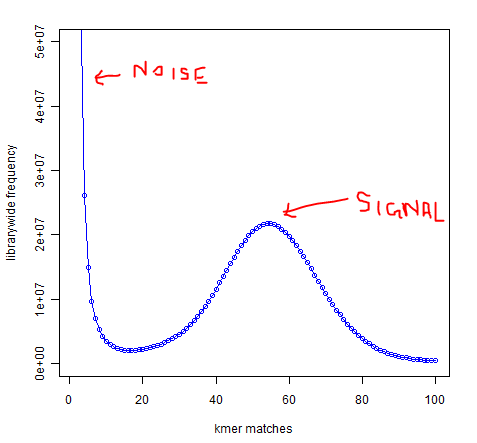
Software such as Quake exists to take advantage of this separation to identify and correct low-frequency k-mers that represent sequencing errors. Removing these low-frequency k-mers should greatly reduce the memory usage of de-Bruijn graph-based assemblers, since every k-mer takes up the same amount of memory regardless of whether it occurs once or 1 billion times.
However, for RNA-Seq transcriptome assembly, the situation is different. Coverage is not even remotely uniform, so one cannot automatically assume a reasonable separation between the noise and signal peaks. Or to put it another way, the Quake website explicitly mentions that it is designed for use with WGS data with a coverage of at least 15x, and in an RNA-Seq experiment, many low-expressed transcripts will probably occur at well below 15x coverage.
So, is k-mer correction like that performed by Quake appropriate as a preprocessing step for RNA-Seq data before running a de-nove assembly with something like Velvet/Oases or Trinity, or is it likely to misidentify k-mers from low-coverage genes are error k-mers and attempt to correct them inappropriately?


I completely agree, but perhaps the poster was thinking about transcriptome assembly, when there might be a greater need for k-mer correction?
Oops, yes, I somehow managed to write that entire question without once writing the word "assembly". I'll edit my question to clarify.