Entering edit mode

14.5 years ago
Jeremy Leipzig
22k
I am using the gbrowse2 samtools setup to display short read data in terms of coverage plots. Gbrowse is selecting different ranges or limits for the plots (as you see below 8, 20, and 50). I want them all to be the same so they can be compared visually.
How do I set this up so that the plots "know about each other"?
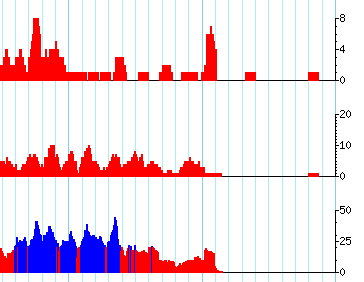
current example of track setup:
[UntreatedReadCoverage]
feature = coverage
glyph = wiggle_xyplot
database = readsUntreated
height = 50
fgcolor = black
bicolor_pivot = 20
pos_color = blue
neg_color = red
key = Untreated read coverage
category = Coverage
label = 0 # Labels on wiggle tracks are redundant.


perl, v5.8.8 built for x86_64-linux-thread-multi Generic Genome Browser version 2.00
Not that it is necessary to answer your question, but can you provide info about Perl/GBrowse versions used? I am after the first bump into Safe::World / Perl incompatibility during GBRowse install.
Thank you very much. There is a new version of GBrowse released today (2.10), and seems that SAM support is one of the main things the authors are trying to improve. I hope to have it installed in the basic setup tonight.