Entering edit mode

9.4 years ago
jhc
★
3.0k
From version 2.3+, The ETE toolkit provides a set of command line tools to perform a variety of tree-related operations (i.e. tree building, tree comparison, visualisation, etc.)
ete build allows to execute complete phylogenetic workflows with a single command line, taking care of all steps from sequence parsing, alignment reconstruction and model selection to tree inference and image generation.
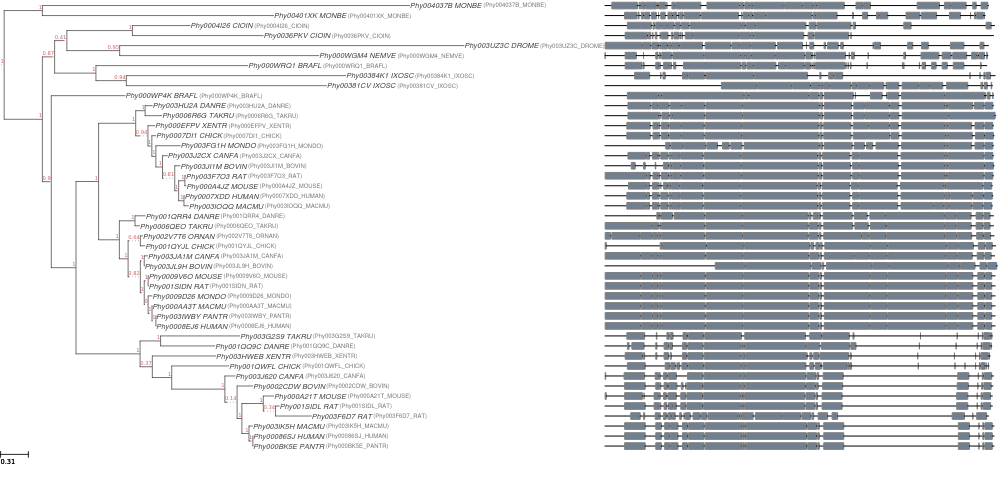
Highlighted features
- Support for both gene tree and super-matrix (concatenated) tree workflows.
- gene family trees require only a fasta file.
- supermatrix-based trees require a fasta file and a COGs file.
- A number of external tools are currently supported (Raxml, Phyml, FastTree, BioNJ, Muscle, ClustalOmega, Mafft, M-Coffee, Trimal, ProtTest); and the framework is prepared to easily implement more bindings in the future.
- The execution of a pipeline can be interrupted/resumed.
- A number of predefined workflows are provided, and others can be created and customised by users.
- Multiple workflows can be executed at once, while all the shared steps will be reused as necessary. This becomes very useful to compare the effect of different aligners or tree inference methodologies, on the same input sequences.
- Pipeline execution is HPC ready
- As the complete tree building pipeline is executed with a single command line, preparation of thousands of jobs and its submission to a computing cluster is piece of cake.
- ETE-build can smartly handle multi core executions by: a) parallelising the execution of independent tasks b) activating multi-threading capabilities of external software (i.e RAxML-PTHREADS).
- NFS friendly. In HPC environments, reading and writing operations on Network File Systems (NFS) can significantly slow down the execution of pipelines. ETE-build allows to specify a scratch directory (i.e
--scratch_dir /tmp), so all the computations will occur on a local disk and results will transparently be moved to the output directory when finished.
- Automatic switching from amino-acid to codon-based nucleotide alignments. If both amino-acid and nucleotide fasta files are provided as input, and a protein similarity threshold is specified, protein alignments will be automatically converted into nucleotides prior to tree reconstruction when necessary.
- Improves reproducibility. The execution of workflows is based on a predefined set to external tools, which are downloaded and compiled from sources automatically by ETE-build. The tool chain is versioned, thus improving reproducibility of results.
The execution of all tasks is monitored:
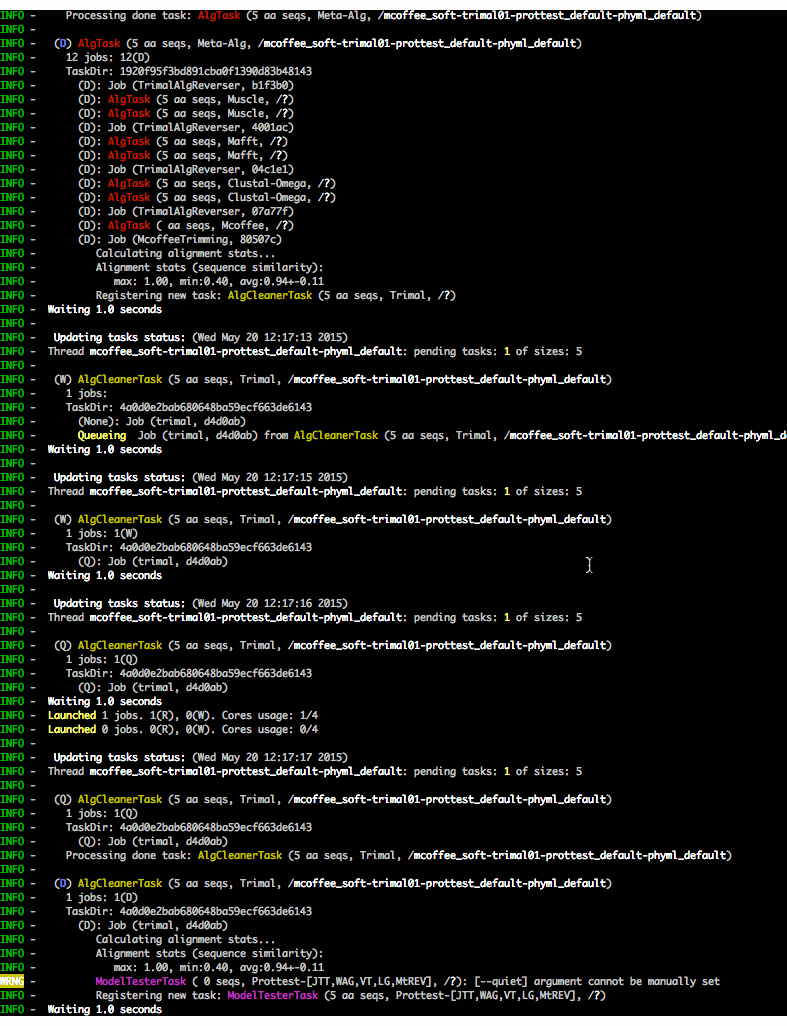
Check ETE-build in action
{kind=link}
Notes:
- At the moment, ETE build is prepared to run only on Linux-based platforms.
- ETE-build uses many external tools that should be acknowledged and accordingly cited. A list of used software and references will be reported after each analysis.
- By default, ETE-build will attempt to generate an image of the resulting tree and alignment. This requires python-qt4 and a X-server. You can disable image generation by using (
--noimg), or simulate and X-server my prepending your ete-build command with "xvfb-run". - ETE-build is a work in progress. Follow the upcoming news or [get involved][10] if you want your favourite tool/workflow to be supported in future releases
We are currently working on getting native support for OSX. We need testers and feedback. Development at: https://github.com/jhcepas/ete/issues/127