Entering edit mode

8.9 years ago
Aurelie MLB
▴
360
Hello,
I apologize if it is a silly question. I am trying to get my head around the Sailfish TPMs.
I can read in the paper that the "estimated abundances are corrected for systematic errors resulting from sequence composition bias and transcript length". But I cannot see anything about the size of the libraries...
Do the sailfish TPM normalize for the size of the library as well please?
Many thanks


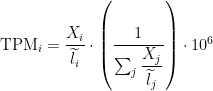 .
.
Hello,
Thanks a lot for your answer. Very helpful!
I did not know I could use the sailfish NumReads for differential expression analysis. I will have a closer look. Thanks!
The first part of your answer is actually the type of explanation I was looking for. So, (please correct me if I am wrong) if I understand well: it should be possible to compare the expression of genes across samples, shouldn't it (even if the libraries are of varying sizes)? I prefer to ask because I just stumbled accross this post where the first post is saying "2. I understand why one shouldn't compare TPM between samples, since the total expression rates, rRNA component etc. varies sample-to-sample.". Well I do not get this very well. I thought that was one of the point of normalising... Sorry I am getting confused now.
Thanks!
So, the reason TPM is not directly comparable across samples is, in large part, because of how aggressively normalized it is. Consider the following (extreme and illustrative) example, you sequence a sample where only 2 transcripts, A and B, are actually present. Say they are present in equal amounts, and there are 5,000 copies of each molecule in the sample. Assuming unbiased sampling and accurate inference, you'll get back TPM estimates of ~500k for each transcript. Now, consider a second sample, where there are 3 transcripts expressed, A, B and C. Further, assume that the number of As and Bs is the same as in our first sample (5,000 copies each), but now C also has 5,000 copies expressed. This time, when we estimate TPM, we'll get a TPM of ~333K for each of A, B and C. This is because the TPM estimates will always sum to 1,000,000. TPM is always breaking up the entire expressed transcriptome into a partition of unity (and then multiplying by 1,000,000). Thus, if the total expression level changes, so will the TPM (i.e. if a transcript is expressed, in "absolute" terms, at the same level across samples, but the expression of the other assayed transcripts changes, then the TPM values of all of the transcripts can change). The normalization approaches used for e.g. DGE, attempt to account for this, by not always forcing the total "expression" to be the same. It's worth noting that it's a tough problem though, because RNA-seq can, by nature, only give you relative estimates of expression (though spike-ins can help one interpret these relative levels). However, TPM, like R/FPKM is designed to be a nice, interpretable unit of expression within a sample, not really to be compared across samples (though you will see people doing this quite often).
Nice follow-up Rob. Even though I knew that TPM normalises for total reads indirectly, I could not have explained to a colleague why that means it is not possible to compare different samples.
I have a question now though :P
Spike-ins are one method to detect total abundances - could another be to do qPCR on a bit of your library (same library that goes on the sequencer, which we may even still have lying around in a freezer somewhere from old sequencing experiments) and get expression of 100 housekeeping genes from each sample. Since qPCR can be quantitative, could this information be used to adjust your TPMs to actual (quantitative) abundances?
Thanks a lot for your explanation!