Entering edit mode

8.7 years ago
Picasa
▴
650
Based on these figures:
http://www.illumina.com/content/dam/illumina-marketing/images/products/truseq_custom_amplicon_workflow.jpg https://sites.google.com/a/brown.edu/bioinformatics-in-biomed/qiime-tutorial-3
I don't understand what is the final structure we got for a paired end fastq:
1) In the second link: a forward read seem to be composed of a barcode sequence and a primer sequence but a reverse read doesn't have a barcode sequence ?
2) If I use Trimmomatic (http://www.usadellab.org/cms/?page=trimmomatic), it says that I can remove adapters but the second link says there is no adapters ??



{kind=link}
Ok but the primer is present in a read file ?
The barcode reads are present in a separate file from R1 and R2.
1) Barcodes and Index are the same ?
In this link, index can be in the same file (in the read id) : https://en.wikipedia.org/wiki/FASTQ_format
Yes and they're usually only in the read ID. While it's possible to include them in a separate file, this is almost never done (it's a sure fire way to both confuse people and break a LOT of automated pipelines).
Not unless the fragment length was shorter than the read length (e.g., when sequencing miRNAs).
So a a read starts always with the target sequence ? (5' position)
99.99% of the time, yes. It occasionally happens that you get adapter dimers on the end. In these cases the adapter sequence will either be soft-clipped (so it doesn't matter) or the read won't align (again, that's OK).
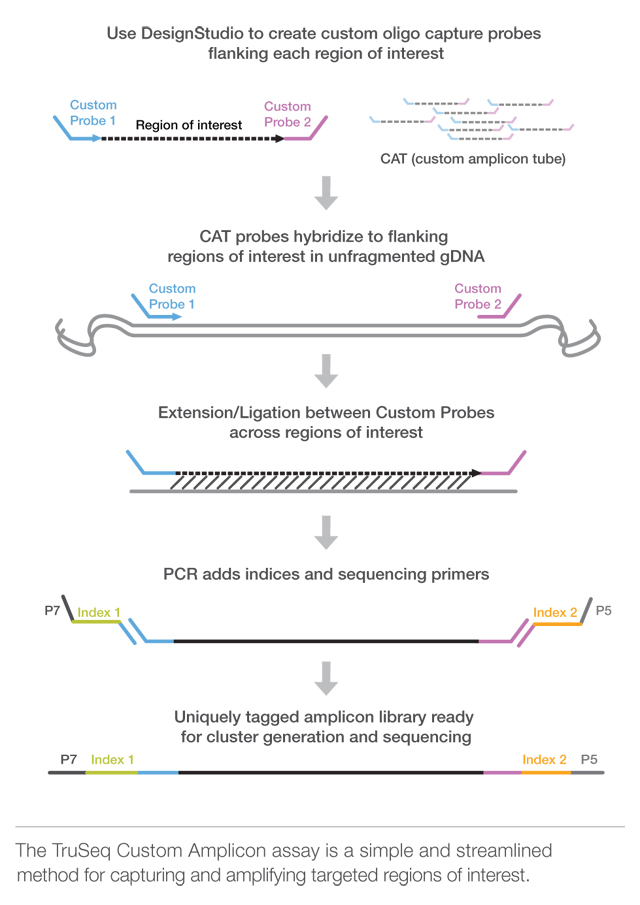
1) So in the first figure: The "Custom probe" is the adapter right ? and what about P7 and P5 ?
2) In the second figure, what they call Primer sequence is in fact the adapter ?
1) So in the first figure: The "Custom probe" is the adapter right ? and what about P7 and P5 ?
Yes. The custom probe is the adapter. The figure is for custom amplicon sequencing. Are you using the same method?
P7 and P5 are the oligomers linked to the read+adapter along with index/barcode by PCR. These sequences attach to the complimentary oligos attached to the illumina flowchip where the actual sequencing occur.
2) In the second figure, what they call Primer sequence is in fact the adapter ?
Looks like that.
The terms primer/adapter are often used interchangeably, as are index/barcode.