
Hello everyone,
I'm pretty new to the field, so forgive me if this is a dumb question. So, I am working on mapping out single reads to the reference mouse genome, with given 260 fastq files. What I'd like to do is make all 260 files to be good quality reads. Right now it's a total mess, as in some have bad 'per base sequence quality' and 'per base sequence content', while some reads have bad 'per base GC content', and so on.
My original thought was run a program that fetches what passed and what failed from summary.txt from each fastqc directory. Then I'd have list of files that failed 'per base sequence content' or failed 'per base GC content' and such. However, because each reads are different within the list, I would still have to open html file and look individually to improve the quality. ( ex. I might have to trim 10 bp while other files need to trim more than that to improve the quality. )
Is there a way to somehow program it so that I can make all the files to be good quality reads at once, rather than checking each fastqc.html files myself and make different adjustments to each reads to improve the quality?


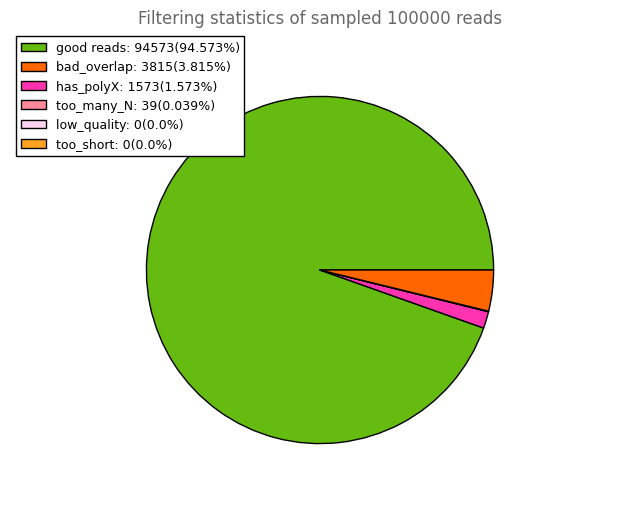 and other subsidiary plots for all the samples for your better understanding. This should be helpful.
and other subsidiary plots for all the samples for your better understanding. This should be helpful.
+1 for MultiQC mentioned below. It can handle logs from several other applications (besides FastQC like aligners etc) and consolidate them in one location.