Entering edit mode

9.0 years ago
ta_awwad
▴
350
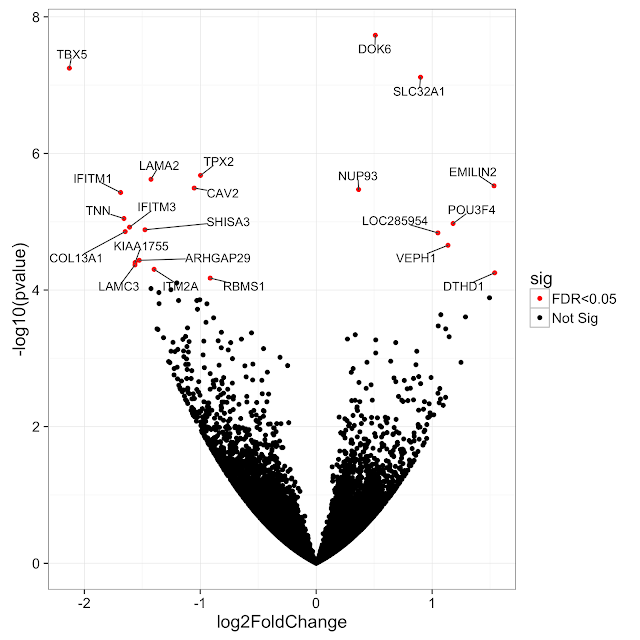
Hello everyone, I would like to have gene names added to volcano plot obtained from DEseq2 ... I have the following matrix:
baseMean log2FoldChange lfcSE stat pvalue padj
Aats-phe 1439.85510 -0.3915108 0.10641530 -3.679084 2.340731e-04 8.682721e-03
achi 1114.41542 -0.4206245 0.10794425 -3.896682 9.751936e-05 4.128319e-03
Act42A 25233.52971 -0.4144380 0.07727588 -5.363096 8.180730e-08 8.283542e-06
Ada 514.03083 -0.6321073 0.13696097 -4.615236 3.926483e-06 2.724179e-04
ade5 3620.63094 0.8756724 0.12531134 6.987974 2.788849e-12 5.804679e-10
Adgf-A 4432.04719 -0.3219797 0.08413694 -3.826854 1.297917e-04 5.173027e-03
alphaTub84B 22505.94872 -0.3424581 0.09110146 -3.759085 1.705361e-04 6.423805e-03
Ama 198.23454 -2.0373321 0.21461357 -9.493026 2.244235e-21 1.401338e-18
Ance 1513.97966 -0.3010685 0.09949477 -3.025973 2.478346e-03 4.876555e-02
as you see DEseq2 doesn't add an identifier to the gene name column (is there any option to do so?) .. and I used the following line to generate volcano plot:
with(subset(res, padj<.05 ), points(log2FoldChange, -log10(pvalue), pch=20, col="red", xlim=c(-10,10))).
now I would like to add gene names to the significantly DE genes.
thanks for your help






Can you append the output of
dput(res)so we can easily recreate the matrix?i would appreciate if you offer a script to do so. I am new in the field ,... that is why i don't want to make any miss