Entering edit mode

8.7 years ago
Sej Modha
5.3k
We have developed an in-house metagenomics pipeline that we apply to the samples when we have sample from unknown hosts or if the host genome sequence is not sequenced. In the absence of the reference genome it is difficult to determine how to remove the host sequences from the sample. The pipeline described here can be applied directly to any virus metagenomics study.
metaViC is available to download from the github.
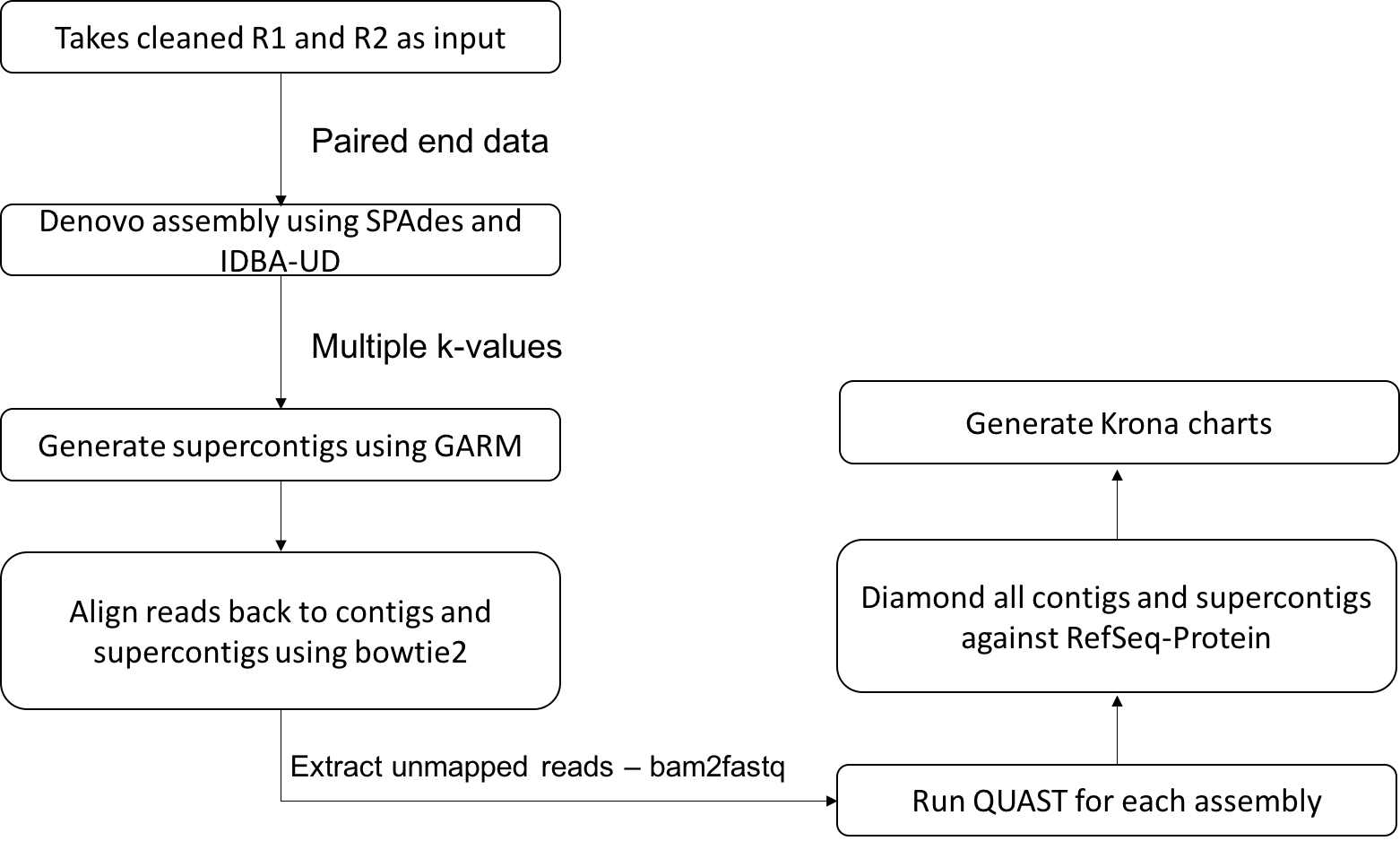
Figures below describes the workflow of the pipeline.
1. Cleaning – removing known higher level organism and bacterial reads

2. De novo assembly
I think it is a brilliant pipeline !
Looks nice. Many of the existing tools claimed to work for this don't do protein-level mapping (with e g Diamond) which is IMO essential.
I agree, especially for the virus metagenomics it is a really important step. Hence in this pipeline we have included a read level and a contig level classification against reference protein database.
This is a really useful pipeline. It would be good to build upon this to more fully characterise the final contigs produced. In my experience, I often end up with contigs that don't match anything at the protein level but match possible hosts at the nucleotide level. Great stuff.
Hello I try to run metaViC, but I have a error
I hope you can help me
Please open a new question, and post the command used to run the pipeline.
...¿y quieres traducir el español al inglés para los demás, Fernando? (...and do you want to translate the Spanish into English for the rest, Fernando?)