Entering edit mode

8.1 years ago
Lars
★
1.1k
Trimming adapter sequences - is it necessary?
Removal of adapter sequences in a process called read trimming, or clipping, is one of the first steps in analyzing NGS data. With more than 30 published adapter trimming tools there is a more than large choice for the appropriate tool. Yet, there is a debate whether this step really is as important as the number of tools suggests, or whether it is possible to skip this time-consuming step for many NGS applications. read more
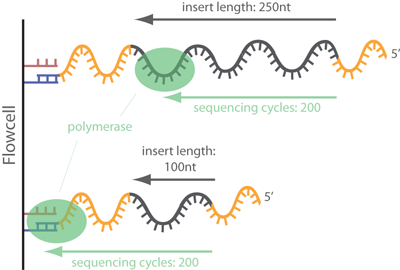




Always best to be safe than be sorry later on.
It all depends what you want to achieve with your data.
Absolutely! But that's what they actually write in the linked text. Not always needed, but sometimes necessary. Nice and simple explanation of adapter contamination. Thanks to the writer.
this used to be a popular thing to do but most people doing resequencing (WES/WGS) don't trim sequences by quality or adapter. see http://gatkforums.broadinstitute.org/gatk/discussion/2957/read-trimming
Sorry for digging out such an old debate, but I was thinking maybe performing a local alignment would replace the trimming of the reads, be it the quality trimming or trimming adapters. Does it make sense? Thank you for any input.
Most NGS aligners are "local" in the sense of the strict-5'/loose-3'.
Bowtie2 for example has an additional option to perform local alignments like this (not a default option)
I think Subread-align works this way as well, but I don't know about other mapping programs.
Most NGS aligners are "local" in the sense of the strict-5'/loose-3'.