Hello All,
When I read this http://thegenomefactory.blogspot.in/2012/09/using-velvet-with-mate-pair-sequences.html ( by Torsten Seemann ), i came across Combining all of them(mate-Paired end, Paired-end and Single End reads) for denovo assembly. I would like to know that why all 3 libraries (SE reads, PE reads and Mate PE reads) of an organism should be combined and assembled? Combining 3 libraries will improve the assembly quality?
That blog post is not a recommendation, it's an example. Just because you can do something, doesn't mean you should. In this case the recommendation is about specifying the input order for Velvet - i.e. smallest insert size first. Generally for a short-read de novo assembly you would use paired-end and mate-pair data only. Adding mate-pair data is definitely a great way of improving an assembly that has been generated from paired-end data.
Hello Daniel,
Thanks for answering. I have two questions for you.
1) You mentioned that "Adding mate-pair data is definitely a great way of improving an assembly that has been generated from paired-end data". In this, did you mean to say that mate-pair data is generated from paired-end data?
2) You mentioned that " Generally for a short-read de novo assembly you would use paired-end and mate-pair data only". In this, why paired-end library and mate-paired end library of an organism should be combined for assembling? Once i get mate-paired end library of an organism, why should not I assemble only mate-paired end library? Should i combine both mate-paired end library and Paired-end library of the same organism to get better assembly results?
When you assemble paired or single reads together, you usually end up obtaining many different contigs that are not connected one to another. This is because you always have gaps, specially when using short reads
Mate paired reads are long distance kind of paired end reads that allow you to order contigs. With mate paired reads you get the end sequences of fragments separated many kb
Let's assume you make mate paired with 10kb long fragments. You get the sequences of both ends after a different protocol involving the auto- ligation of the 10kb fragments. A search in Google will give you details
If you find the left mate paired in contig #1, and the right mate paired in contig #400 (and this is an example), this means that both sequences are separated 10 kb.
This allows you to order contigs and find their corresponding neighbors
Hello,
Thanks for answering. But i am still not clear. My question is that why paired-end library and mate-paired end library of an organism should be combined for assembling? Once i get mate-paired end library of an organism, why should not I assemble only mate-paired end library? Should i combine both mate-paired end library and Paired-end library of the same organism to get better assembly results?
If you assemble with paired-end reads only you ALWAYS end with many different contigs. You lack information about how to connect them or in which order they are in the genome. Gaps are almost always present in assemblies
This should be easy to figure out..
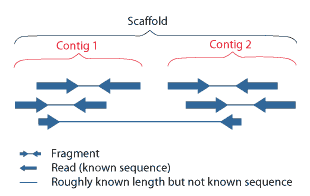
With the mate-paired sequences you help in the assembly because you are increasing the coverage in first place, but more importantly is that you can connect or find a relationship among the contigs and at the same time you introduce a length information in your genome sequence. This is scaffolding..
Maybe these pictures can help you in understanding this images
Yes you combine them both. The mate-pair data allows you to do things like assemble across repetitive regions that you cannot resolve with paired end data. You wont get very far assembling mate pair data alone, and if you look at how mate pair libraries are generated you will see why.
Hello Daniel,
In this paper "A field guide to whole-genome sequencing, assembly and annotation", I have read the following point that "After the initial contig building, it is common to use read-pair information from long-insert (mate-pair,fosmid-end or jump) libraries to combine contigs into scaffolds". So i would like to know that the mate-pair library which is combined with paired-end library will help in generating better/lengthier contigs or help in combining contigs into scaffolds?
Mater-pair information has a high level of duplication. And the reason you need mate-pair is only for arranging fragmented blocks based on distances (imagine mile markers). If you need to actually assemble data, you need information based on adjacencies, that is what you get from paired-end information. Even though you get blocks from adjacencies, they are broken either due to repeat content or genome complexity or lower coverage regions. This is where our mile-markers of mate-pairs come in for arranging continuous blocks together.
Yes you need to combine both for more information as paired-end gives you fragmented blocks and mate-pair the distance estimates, which need to be put together.



Hello Daniel, Thanks for answering. I have two questions for you.
When you assemble paired or single reads together, you usually end up obtaining many different contigs that are not connected one to another. This is because you always have gaps, specially when using short reads
Mate paired reads are long distance kind of paired end reads that allow you to order contigs. With mate paired reads you get the end sequences of fragments separated many kb
Let's assume you make mate paired with 10kb long fragments. You get the sequences of both ends after a different protocol involving the auto- ligation of the 10kb fragments. A search in Google will give you details
If you find the left mate paired in contig #1, and the right mate paired in contig #400 (and this is an example), this means that both sequences are separated 10 kb.
This allows you to order contigs and find their corresponding neighbors
Hello, Thanks for answering. But i am still not clear. My question is that why paired-end library and mate-paired end library of an organism should be combined for assembling? Once i get mate-paired end library of an organism, why should not I assemble only mate-paired end library? Should i combine both mate-paired end library and Paired-end library of the same organism to get better assembly results?
Ok.. Read carefully
This should be easy to figure out..
Maybe these pictures can help you in understanding this images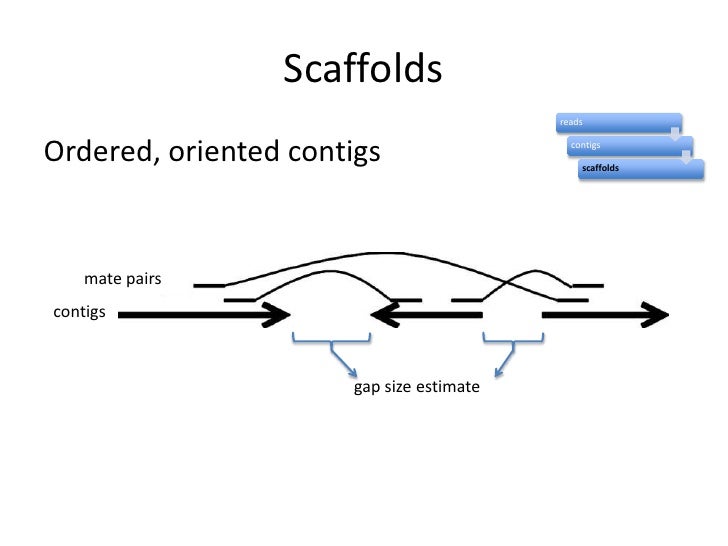
Thanks a lot for clearing my doubt.
Yes you combine them both. The mate-pair data allows you to do things like assemble across repetitive regions that you cannot resolve with paired end data. You wont get very far assembling mate pair data alone, and if you look at how mate pair libraries are generated you will see why.
Hello Daniel, In this paper "A field guide to whole-genome sequencing, assembly and annotation", I have read the following point that "After the initial contig building, it is common to use read-pair information from long-insert (mate-pair,fosmid-end or jump) libraries to combine contigs into scaffolds". So i would like to know that the mate-pair library which is combined with paired-end library will help in generating better/lengthier contigs or help in combining contigs into scaffolds?
I think I did not clear your doubts...
Mate paired will mainly contribute to scaffolding.
However they also contribute with reads so they can also influence the formation of contigs, but this is not their main advantage
Thanks for answering again.
I think you would benefit from our de novo Assembly course
http://earlham.ac.uk/de-novo-assembly-2017