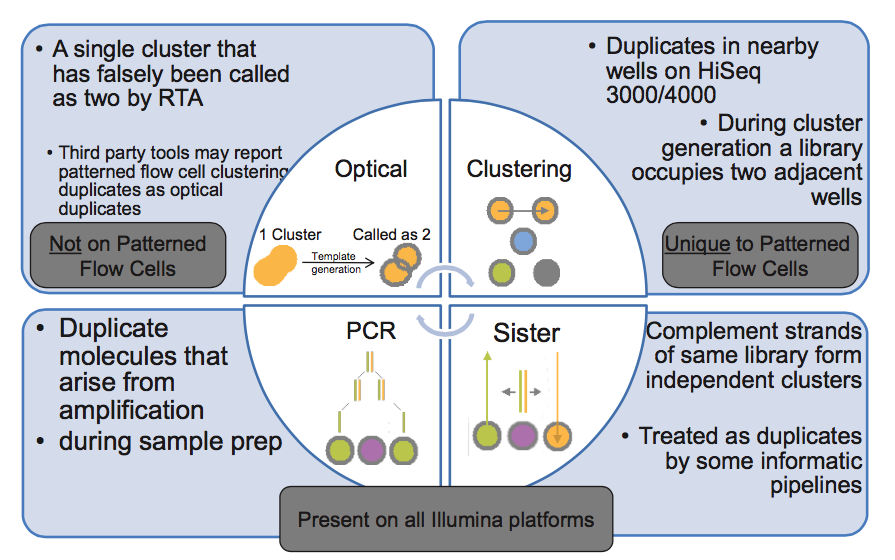
I've come across this picture from Illumina presentation explaining how duplicate reads can happen. Two types of optical duplicates and PCR duplicates are pretty clear to me, but lower right corner has me confused. Could anybody explain what are these "sister" duplicates?
On a related note, what happens to the second strand when during the cluster generation the fragments are denatured with 2M base? Does it just not attach to the flow cell and is later washed out?
Does not it mean, that if you have double stranded (complement strands), when each strand is create independent cluster position. So after sequencing you have the same data set but in reverse/complement orientations, but after alignment to reference is the same starting position = flag as duplicates?
Well I don't think something like Picard markduplicates would mark it as a duplicate if sequence is the same but the strand flag is different. Need to check it to be sure.
Correct - reads that map to the same position but on opposite strands are not marked as duplicates by Picard, nor are they removed by SAMtools rmdup command. But those reads would have to be from independent library molecules (i.e., not duplicates). Read one always proceeds from the same adapter end (I believe it's P5) so, even if sister clusters were formed from the two strands of a single library molecule, their orientations relative to P5 would be identical.
I don't get it. You would have P5 on one strand and P5's complement on the other, would you not? And the complement should not be able to attach to the flow cell.
You're conflating two issues. My point was that, if two reads have opposite orientations, they cannot be duplicates of any type (PCR, sister, or optical) b/c all inserts are sequenced relative to the same adapter end and strand.
Regarding annealing, the flow cell contains both P5 and P7 anchor primers. These serve as PCR primers for bridge amplification (aka clustering). If one strand of the library anneals to P5 (i.e., is the reverse complement), then the other end of the other strand MUST be the reverse complement of P7 (draw a diagram if this is unclear). But one of the two anchor primers is blocked during annealing (see discussion below), which is why sister duplicates are not produced.
I've identified another type of duplicates not on this chart: tile-edge duplicates. These appear to be caused by the camera not sliding over far enough when moving to a new tile. In my analysis of NextSeq data, these account for the vast majority of duplicates (>80%). The picture shows red dots for unique clusters and blue dots for duplicate clusters, and the X/Y axis are flowcell coordinates. All 13000 duplicate pairs are plotted for the first 9 tiles of this NextSeq run; the non-duplicates were subsampled to 13000 before plotting. These duplicates were detected via Clumpify rather than mapping.
Note that in this image, all 9 tiles are superimposed on top of each other (the X/Y coordinates get reset for each tile). Also, from what I have read, the NextSeq runs 3 tiles wide on a lane. So, I would expect duplicates on both sides of the middle column tiles; right side only for the left tiles; and left side only for the right tiles. Thus, the presence of nonduplicate reads on the edges (at a much lower rate than elsewhere) is probably the contribution from left and right tiles.
I think this is related to the large cluster size (they are on the order of mm in diamter, if I recall correctly, compared to other Illumina sequencers) for NextSeq. I have only tried a couple of samples (from HiSeq 4K and MiSeq) and have not seen this.
Hahahah - oh boy. Fantastic research as always, but wow, what to say.
I guess they wont be marked as optical-duplicates with tools like Picard as they are thought to come from different tiles? I mean, it will detect all duplicates regardless, but detect as optical duplicates.
I've only tested our NextSeq, so I don't know if it's universal or ours is miscalibrated, but we will be discussing it with Illumina soon. Seems like it should be an easy fix. In the meantime I'm updating BBDuk to support location filtering so we can exclude all tile-edge reads.
That will give you 20000 randomly-sampled duplicate read headers in "duplicates1.txt" and nonduplicates in "unique1.txt". I pasted that into Excel splitting on spaces and colons. The headers look like this:
So the X and Y coordinates are 20306 and 1857. You can, if you want, just start with the first 2 million reads to speed things up a lot and use less memory, if you have a huge file.
Hi Brian Bushnell, a bit later than I planned but I made some plots...
Looks like we have the same issue. Can you keep me posted about your correspondence with Illumina?
Plotting code:
This file contains hidden or bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
Thanks Wouter! I'll keep you informed. Actually, I think it would be fun to post Illumina's first response:
Hi all, I reviewed the run to see if there are any issues related to
a subset or all of the 6 cameras on the instrument by looking at
sequence quality overall as well as per camera section to gauge the
variability.
On the whole, the run itself performed well by exceeding our quality
specs for a 2x150 run: 85.6% vs 75% spec. All 6 cameras are also
performing to spec, the variability is 6.8%, where we would expect
<20%. For more information on camera variability in general, please
refer to the attached technical note, and data specific to this run
can be found in the second PDF attachment.
In summary, it looks like the system is imaging as expected and does
not need calibration at this time.
So - yeah. Looks like typical Illumina quality control. I did forward them some images in response that, who knows, maybe they didn't get initially... hopefully their next response will be more useful.
Thanks for sharing. Illumina then has a quite limited idea of "quality" and "as expected"... I'll inform those responsible for our sequencers on Monday.
Hi Brian Bushnell, I'm not sure how the tiles are organized on mid output flow cell, but it's likely different.
Have you investigated if the edges have overrepresented sequences or only looked at duplicates?
I've only looked at duplicates. Note, though, that you can use BBDuk's new positional filter (xmin ymin xmax ymax flags) to select reads in a rectangular region, if you want to run FastQC on just a subset.
I selected the edges using BBDuk with everything left from x=2000 and everything right from x=25000 and compared the fastQC report from this set with the all the reads, and those are very similar, nothing odd.
Legally they are in the clear. NextSeq specs are listed on this page. Only criteria are % of reads with Q30 and number of reads passing quality filter. That is pretty much the norm for all Illumina sequencers.
I'm going to look at a few more runs to judge run-to-run variability.
As an aside, I'm trying to come up with a better visualization. I'd prefer a density plot of some sort, but they keep producing edge-effects that counteract what's otherwise visually evident.
I was thinking of density plots on X and Y axis, expecting to see the duplicates-effect when collapsing the data to X and expecting to see nothing special when collapsing the data to the Y chromosome. But that isn't showing the desired data?
Could it be that the run you processed already contained a lot of duplicates, therefore relatively decreasing the effect of the tile-edge duplicates?
I've settled on binning both axes and then plotting the % duplication in each, since otherwise the read density tends to skew the density plots. This shows a nice uptick at the edges.
Brian Bushnell is allowing 5 substitutions in this clumpify.sh command line (per read) so keep that in mind. Technically those are not optical duplicates.
NextSeq clusters being huge may be a completely different beast. Brian Bushnell I will take a look at this on the HiSeq 4K data we were looking at.
FYI, in version 36.81 of clumpify, the allduplicates option only works if groups=1 (or you use small enough files that it sticks everything in memory by default). I assume this is a bug.
I would expect them to not be identified as optical duplicates by Picard because they are on different tiles and their coordinates are different by at least 25000 pixels on the X axis. They should still be identified as duplicates, though, since they would map to the same location. The HiSeq 4000 data I have seen so far does not show this problem.
I have not heard anything about a fix, and Illumina's suggestion for a long-term solution when we discussed it with them last was simply to cut off the leftmost and rightmost X pixels from every tile; there was no indication that they planned to change anything.
I (just now) tested a NextSeq run from last week to check, and it still shows the same problem. It's possible that you are using a newer version of Illumina's software that fixed it, though. How did you test it to determine the problem appears to be solved?
I screen all NextSeq runs we do as part of our standard pipeline and typically find some small portion of duplicates (the one that finished last night had up to 1%, depending on the sample). So no, the issue hasn't gone away.
This file contains hidden or bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
I then plotted the last output file (read_kinds_sampled.txt) using the R script shared earlier in this thread.
Some of our recent NextSeq runs had samples with 140 million 2x150bp read pairs, so using mostly stream-based tools and downsampling to 2 million reads made this analysis faster.
Are your NextSeq's newish (recently purchased)? Older sequencers tend to have issues that may be fixed in newer iterations. Illumina may not always advertise changes they make internally to the hardware.
One NextSeq was new, I believe, in March 2016 and we just got a second one 1-2 months ago that was used. Not sure exactly how old that one is (I will try and find out)
I don't recommend downsampling when doing positional duplicate analysis. Instead, if the full dataset is too big, just use the first 2m reads instead of 2m random reads (as long as you get reads from at least 4 tiles you should have enough).
Also, note that the duplicate rate in this analysis drops at the edges of the tiles. This is because the quality decreases as the tile goes out of focus away from the center, so duplicates are harder to detect. It's useful to allow one or two mismatches when doing duplicate detection, for this reason, particularly when you expect duplicates to be at the edges. If you have 151bp reads, you may also want to trim them to 150bp first, as base 151 tends to be very low quality.
Usually, the molecules sent for sequencing are double-stranded DNA. But in the procedure for loading the sequencer, the DNA is denatured. Therefore, for one double-stranded molecule sent for sequencing, two reverse-complementary molecules are loaded in the machine. Each of them can form a cluster, and these clusters will be indistinguishable. Thus, even in libraries prepared without any amplification step, the sequencing results can produce two identical reads. If the goal of the sequencing is to count molecules (transcriptome sequencing, single-cell epigenome analysis, ...), it is better to count these "sister" sequences as one.
The way I understand flowcell attachment works, only one strand should be able to attach and form a cluster. I might be wrong though.. so the other strand is attaching with P7?
When I first wrote a duplicate-removal program, I was worried about the possible presence of "sister" duplicates, so I designed it to detect both identical and reverse-complementary duplicates. I was not concerned about the presence of these duplicates because DNA is double-stranded and thus a fragment might spawn two clusters - in fact, I doubt that's likely. I assume that when DNA is fragmented, the two strands will generally break independently. Rather, I was concerned that during PCR, a single fragment might spawn both identical and reverse-complementary fragments, so both would need to be removed.
However, in testing, I found that the rate of identical fragments was high, and the rate of reverse-complementary fragments was extremely low - a rate that could be completely accounted for by coincidence. So, I don't worry about it any more. Though I should mention that I don't really understand why this is the case - for a PCR-amplified library it still seems to me that reverse-complementary duplicates should be present. Perhaps someone else has some insight here?
Illumina always shows fragments clustering in one color (orientation) in illustrations. Possibly the other class of anchors are blocked (until made available during the turnaround phase)?
I imagine so - but as always a few reads might not get blocked every now and again. Or perhaps it's a very different situation, where the denaturing of the original template occurs after the entire flowcell has been flooded with dsDNA. I imagine if you flooded a flowcell with ssDNA, there would be far more clusters at the top of the lane than the bottom. So they probably keep things dsDNA until the flowcell is covered, then denature. This would, and i'm just guessing here, give a more even cluster formation across the lane, and sister fragments would likely also bind to the same physical location as each other. Thus, if they do form independent clusters, they're likely to become merged. Only if sisters travel a significant distance apart after denaturation can they become two independent clusters.Thats just a total guess though. Someone needs to look at the actual protocol :P
@John, the library is denatured with NaOH (and neutralized in hyb buffer) before loading onto the instrument. And yes, there is usually a cluster density gradient (high -> low) across the flow cell from inflow to outflow.
P.S.-I checked my notes from an old (circa 2011) Illumina presentation; as @genomax2 suggested, one of the two primers on the flow cell is blocked during library loading.
Ah, awesome, great info Harold - in that case the mystery is solved. I guess only a small number of sisters are seen because only a small number make it past the block, or aren't fully washed out after the block is removed.
Off-topic, but do you happen to know why flowcells can't be reused? I've always thought that adding an exonuclease to cut off the synthesised fragment at the end would essentially reset a flowcell to being just a lawn of adapters as before. The exo to do that is used in step 3 or 4 of the protocol (after bridge amplification in the first round of sequencing to get everything homogenous 5' -> 3'), so i'm surprised i haven't heard stories of people at least trying it out, particularly since flowcells are $25,000 each or whathaveyou.
Flow cells aren't quite THAT expensive (more like $6K for the current PE version, and that includes reagents). Plus, we all know that the real expense of NGS is the bioinformaticist's time ;-).
The flow cells can't be reused because the anchor primers are cleaved after clustering. Both of the anchor primers are chemically modified to allow differential cleavage. After initial library loading/clustering, primer A is cleaved in the middle so that only the strand tethered to primer B remains as a template for sequencing. Upon completion of read 1 and index reading, the stub of primer A is used to synthesize the complement of the primer B-tethered strand. Then, primer B is cleaved, leaving only the newly synthesized strand as template for read 2.
Ah, see, this is the difference between watching the videos, and knowing the science. Thanks again Harold :) Also, word to the wise, do not Google Image search "differential cleavage" in a Starbucks. You'll get weird looks.
According to the supplemental material of Bentley et al., 2008, clusters in single-read flow cells were linearised with a chemical method, but the ones in paired-end read flow cells were linearised using enzymes related to base excision repair mechanisms, which I doubt will function on the anchor primers when they are still single-stranded. Thus, it might be possible to reuse the paired-end flow cells. I do not know if this still holds true on Illumina sequencers using ExAmp instead of bridge PCR to generate clusters (for instance HiSeq 4000).
Cleavage occurs after clustering, and the anchor primers serve as templates for the clustering PCR, so they're not single-stranded during the cleavage reactions. And whatever they use for cleavage clearly works. Unless it's some offset restriction enzyme like SapI, the cluster/cleavage cycle removes the modification from the anchor primer, so it's hard to envision a method for removing the read 2 template strand and precisely regenerating the two anchor primers for reuse.
Yes, the cleavage is done on the double-stranded DNA that makes the clusters. But I thought that the question was whether there would be single-stranded primers left for re-using the flow cell ? In that case, the point I want to make is that the unreacted primers in the 2008 PE/bridge-PCR design may still be pristine and available for a new clustering reaction.
@John's original question was whether exo (or similar) treatment would regenerate a pristine flow cell, so I responded to that question. But I can envision a few problems even in your scenario of unreacted primers:
1) Strand 2 of the first run would remain, and would be a template for read 2 of the second run, which would invariably affect base-calling of adjacent run 2 clusters (and probably S/N metrics as well, since they're calculated independently for each read).
2) The block to sister duplicate formation would be missing, so ~half of the data would be redundant.
3) The loading capacity (cluster density, number of reads) of the flow cell would be significantly reduced.
While that may have been true in 2008 there has been a change to this since. If I recall, now both SE/PE flowcells use a similar mechanism. @harold: does this ring a bell by any chance?
MiSeq (and I believe NextSeq) kits work for both SR and PE sequencing. But the HiSeq and Rapid kits come in both SR and PE versions, and the flow cells are not interchangeable. Of course, you could always perform SE sequencing with the PE kit but, given the substantial difference in price, why would you want to?
But this thread has drifted far from bioinformatics, so I'm signing off. Additional queries on this topic should be posted on SEQanswers.
Just not worth the hassle, even if you could do it. Plus illumina sells them as a part of a kit so there is no way to just buy the reagents (no third-party suppliers).
People are worried about getting cross contamination of reads from different tags in a pool and you are wanting to re-use a flowcell :)
I have no idea what all these colours are about, but i think they're saying that it is possible for both strands from a single DNA molecule to form separate clusters on the flowcell. In the Illumina "how its made" videos there's always only 1 strand of DNA annealing to the adapter lawn and kicking off bridge amplification. I guess in reality the other strand could also anneal, and thus 1 DNA molecule could form 2 clusters that would appear to be PCR duplicates, but actually are just "sisters".
Yeah i'm not sure how serious an issue that is. Perhaps people with barcoded, non-amplified samples saw the same fragment sequenced twice and freaked out, then it was realised that this is an actual thing that happens. Otherwise you wouldn't be able to tell it from standard PCR duplication.
Shouldn't be technically possible, right? You would only have a correct P5 sequence on one strand, the other one would have a complement. That's what I am confused about.















Does not it mean, that if you have double stranded (complement strands), when each strand is create independent cluster position. So after sequencing you have the same data set but in reverse/complement orientations, but after alignment to reference is the same starting position = flag as duplicates?
Well I don't think something like Picard markduplicates would mark it as a duplicate if sequence is the same but the strand flag is different. Need to check it to be sure.
Correct - reads that map to the same position but on opposite strands are not marked as duplicates by Picard, nor are they removed by SAMtools rmdup command. But those reads would have to be from independent library molecules (i.e., not duplicates). Read one always proceeds from the same adapter end (I believe it's P5) so, even if sister clusters were formed from the two strands of a single library molecule, their orientations relative to P5 would be identical.
I don't get it. You would have P5 on one strand and P5's complement on the other, would you not? And the complement should not be able to attach to the flow cell.
You're conflating two issues. My point was that, if two reads have opposite orientations, they cannot be duplicates of any type (PCR, sister, or optical) b/c all inserts are sequenced relative to the same adapter end and strand.
Regarding annealing, the flow cell contains both P5 and P7 anchor primers. These serve as PCR primers for bridge amplification (aka clustering). If one strand of the library anneals to P5 (i.e., is the reverse complement), then the other end of the other strand MUST be the reverse complement of P7 (draw a diagram if this is unclear). But one of the two anchor primers is blocked during annealing (see discussion below), which is why sister duplicates are not produced.
Got it, thank you for the explanation.
A blog post has appeared discussing HiSeq 4000 duplicates from Babraham group: https://sequencing.qcfail.com/articles/illumina-patterned-flow-cells-generate-duplicated-sequences
This has also been discussed extensively in this SeqAnswers thread: http://seqanswers.com/forums/showthread.php?t=72901