Entering edit mode

8.4 years ago
colin.kern
★
1.1k
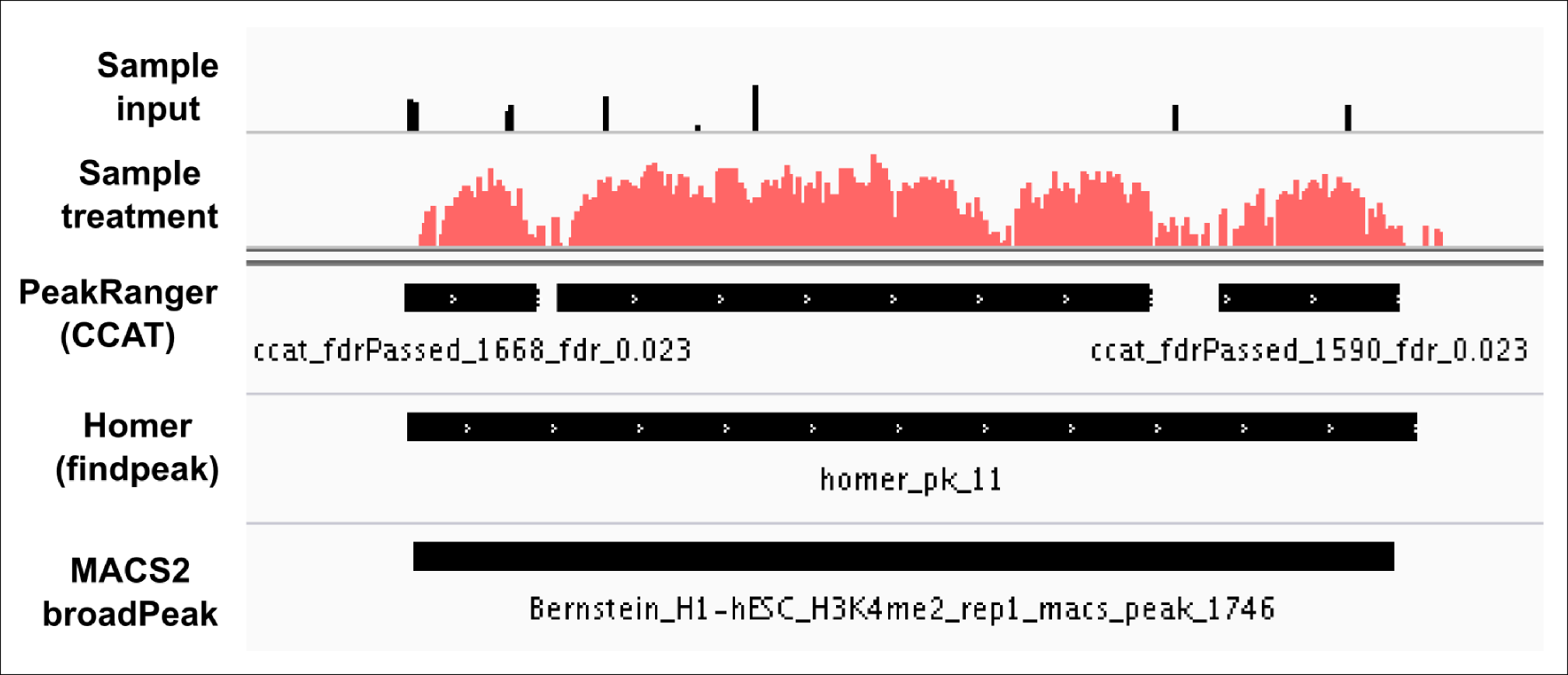
I've noticed that most of the major projects involving wide-scale ChIP-seq analysis like ENCODE and Blueprint use a pipeline that involves running the SPP peak caller to estimate fragment size, then using Macs2 to do the peak calling but with the "--no-model" flag and giving Macs the fragment size estimated by SPP. Does anyone know the reasoning behind this? Is there a paper that shows the method used by SPP to estimate fragment size (cross-correlation) is more accurate than what Macs2 does? And how big of an impact does this have on the peak calling?