Is there a better explanation than just imperfections in processing pipelines or annotations ?
The reason I ask is that I would like to know if there is a lower limit of first exons at the time of RNA splicing. (And the reason behind the reason is that I have a sequence library from which I can infer the length of first exons, and I would like to test if the results are reliable).
Thanks Mark for the detailed answer. And thanks as well to Sanger's help desks who also always gave me prompt and detailed answers. I hope that this discussion on Biostars can be useful to others, which is why I opened it in parallel.
Back to 5′ ends, is there a way to infer from GENCODE's GFF files whether a given transcript model may be truncated ?
It is only possible to directly identify 5' truncated transcripts from the GFF, if the CDS is also truncated, which are identified using the 'cds_start_NF' tag (NF; not found). You may also want to consider using the GENCODE 'basic' gene set, as this prioritises 'full-length' coding transcripts or the longest noncoding transcript isoforms.
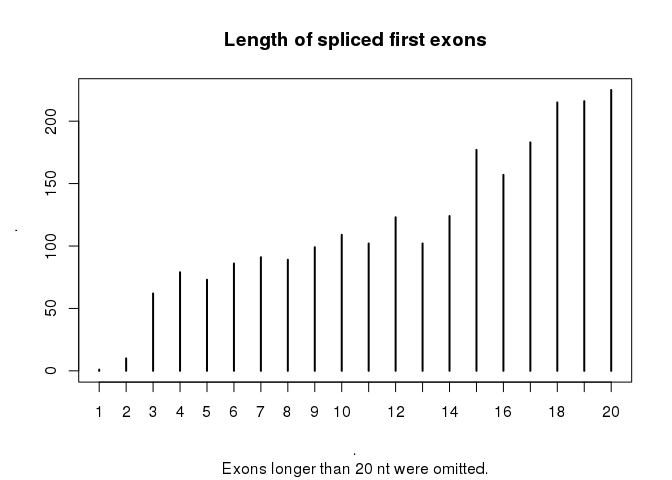
This is an interesting analysis of the GENCODE v26 geneset, that highlights potential idiosyncrasies in the annotation process. There are however some important points to consider when interpreting these results.
While the current geneset contains first 'exons' with less than 20nt, these do not necessarily represent the full-length of the exon. Indeed, these first exons may not actually represent the transcriptional start of the associated transcript. This is because manually annotated GENCODE transcripts are extended to the limit of the supporting evidence, which typically consists of ESTs and cDNAs. If this evidence doesn't extend to the actual TSS, then the annotated transcript will essentially be truncated at the 5' end. This appears to be the case for most of the 'micro first exons' identified here, as they coincide with longer exons in other transcript isoforms for the same gene.
Although the annotation of such short exons may be accurate, their actual value within the geneset may be limited. It may be best therefore to exclude such transcripts from any analysis of RNA splicing. On behalf of GENCODE, we welcome any comments or questions, which can be sent to either HAVANA or GENCODE.


Thanks Mark for the detailed answer. And thanks as well to Sanger's help desks who also always gave me prompt and detailed answers. I hope that this discussion on Biostars can be useful to others, which is why I opened it in parallel.
Back to 5′ ends, is there a way to infer from GENCODE's GFF files whether a given transcript model may be truncated ?
It is only possible to directly identify 5' truncated transcripts from the GFF, if the CDS is also truncated, which are identified using the 'cds_start_NF' tag (NF; not found). You may also want to consider using the GENCODE 'basic' gene set, as this prioritises 'full-length' coding transcripts or the longest noncoding transcript isoforms.