
Hello everyone,
Here's my problem: I have multiple VCF outputs that I have obtained using MuTect2 for 18 patients in total from Whole Exome Sequencing (WES/WXS) (so 18 VCF files). I have a BED file which has the list of all genes. FYI, MuTect2 reports single variants and INDELS in the VCF.
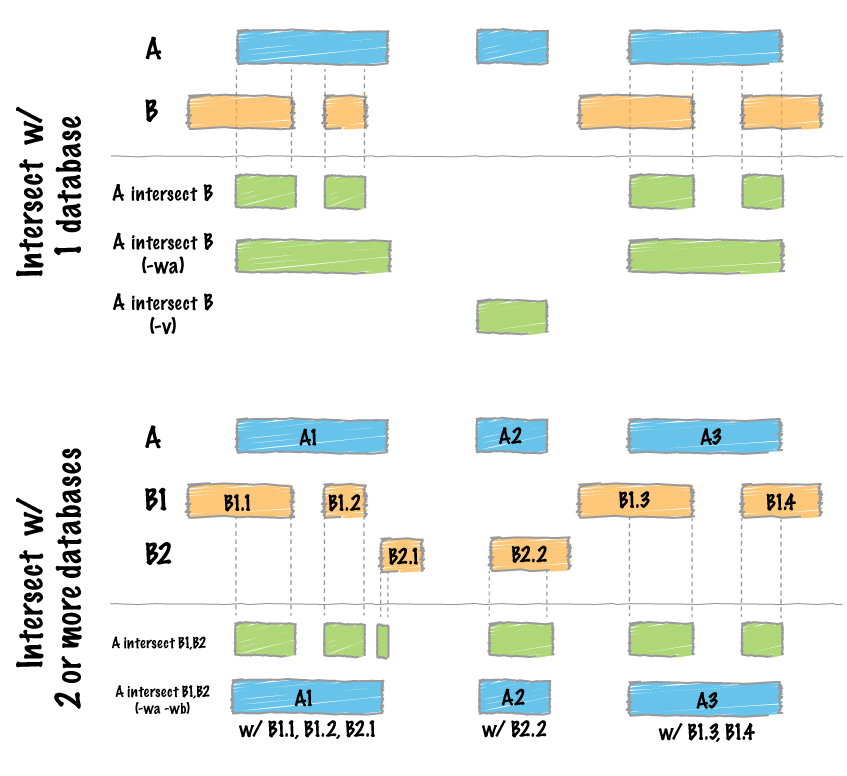
So, if I use bedtools intersect, it report each feature in A that overlaps B, even if its only once. What I want is to do an intersection between the list of genes in BED and all the VCF outputs, for example, at least 5 VCF that reports the same gene in the region. Bedtools describe here:
if I use:
intersectBed -wa -a [BED] -b [all VCFs]
It will report the list of all genes where the variant has been seen at least once, for example:

In the previous figure, you can see that bedtools (BT) will report gene B but I do not want that. What I want is that bedtools report gene A only if 5 VCF (or more) that have variants/INDELS overlap the same gene.
Is there any way to do it ? If there are other tools that you can suggest also, please let me know. Thank you in advance !
Alaa

@Alex Reynolds
Thanks for the help ! I will try it asap !