Entering edit mode

7.8 years ago
wenhui
•
0
Hi, everyone.
I used Tophat2 + cufflinks +cuffmerge to process my RNA-seq data. The annotation file I used is UCSC Hg38. I saw some strange outcomes form the merged gtf file. There are lots of transcript starting at 0 and end at 1. It really confuses me. Should I keep these data or just trim them?
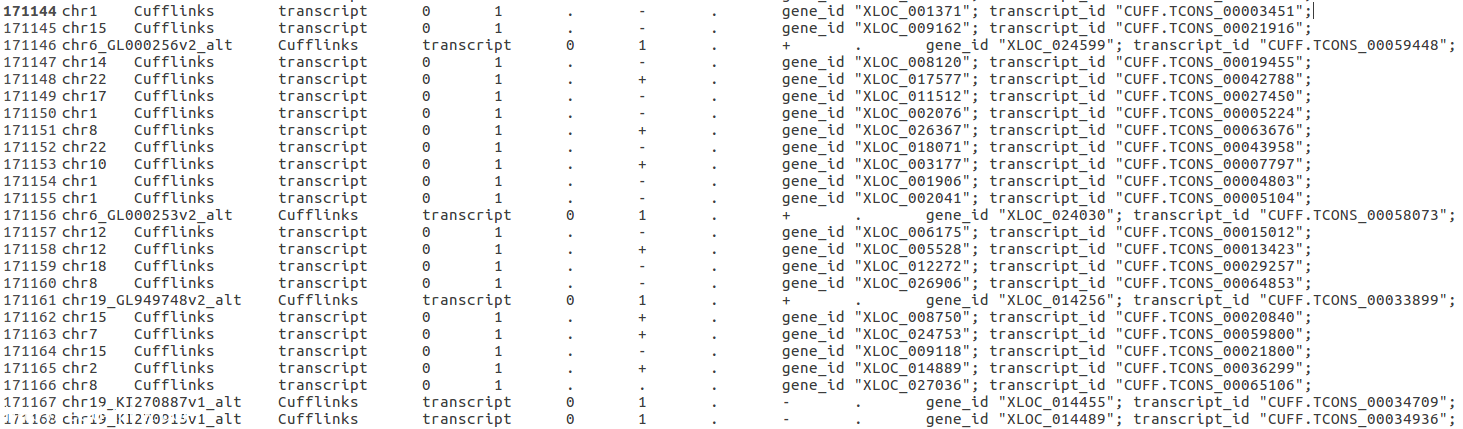
Any advice is appreciated.
Thank you.

Thank you for your reply. I have tried hisat2 +stringtie but the software I used for downstream analysis only support gtf file output from cufflinks. It may have something to do with some prefix in cufflinks' gtf file.
Usually, I prefer hisat2 + stringtie in my work and I found it is much faster than Tophat2 + Cufflinks.
Thank you anyway.