When Salmon (v-0.8.2) is run on each strand of an annotation region separately (given IU as my library type), why wouldn't the TPMs be the same for each strand? From my knowledge of how read-pairs are counted, and that the library type is unstranded (yes I checked the GEO info) shouldn't the TPMS be the same...
Image from sailfish docs
This is actually not _too_ surprising. Specifically, what you're losing in this case is the actual pairing information. Often, a pair of reads is more informative than either of the individual reads, and so the read pair maps to a more restrictive set of transcripts, and hence leads to more accurate quantification estimates. It's also the case that with single end reads, you must set the fragment length distribution explicitly (or let Salmon use it's defaults, which you should only do if you don't have the relevant information for your particular sample available). These differences in the observed fragment length distribution will also lead to different inferred effective lengths for the transcripts (and hence a different maximum likelihood abundance distribution). In short, I'd expect there to be reasonable correlation between the paired-end estimates and those from the left and right reads individually, but I would not expect them to be the same (or even very highly correlated) because of what you lose going from paired-end to single-end reads.

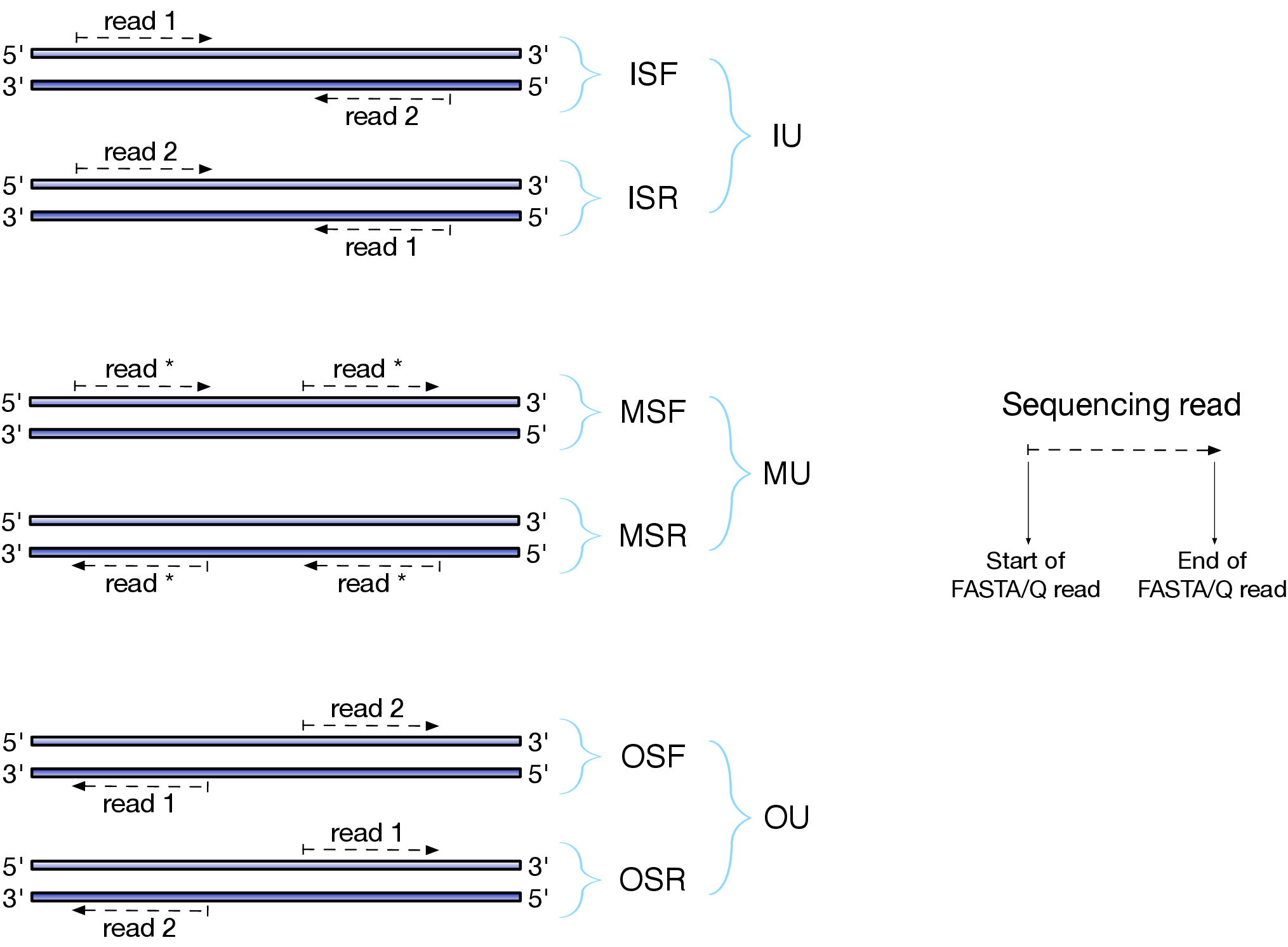 Image from sailfish docs
Image from sailfish docs