Entering edit mode

7.7 years ago
cjgunase
▴
50
Hi All, I am a newbie to sequencing technologies.
Is there a way to quantify the paired-end sequencing overlap. since completely overlapping the read pairs would be a waste of sequencing resources. A bit of overlap can be useful when doing the alignment but a small gap is optimal to maximize coverage.
Is there a way to check sequencing efficiency by using the alignment files. Because if there is lots of overlap we want to improve this.
Any help is appreciated.
Thank you.

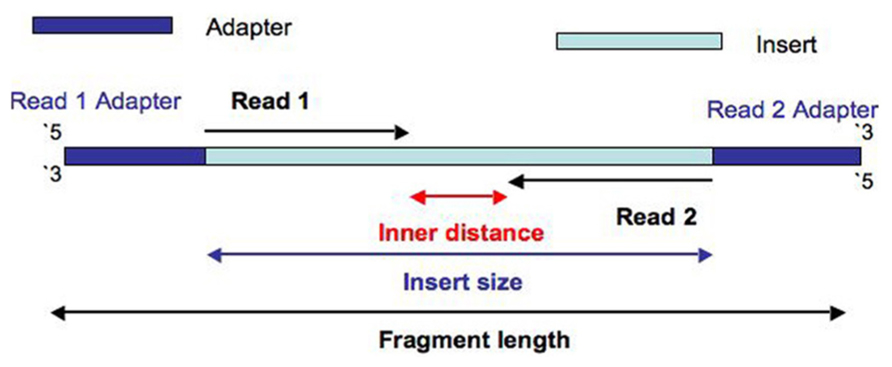


no agree: If the two reads overlap, it usually means that the sequenced fragment was too short.
in paired-end sequencing, you'd better consider the sequencing depth to "maximize coverage".