
Hi,
I had a couple questions about some RNA-Seq data. A colleague recently received some RNA-Seq data and we are attempting to trim the data. We are having some issues with the "per base sequencing content" and the "Sequence length distribution". I attached some pictures of the FastQC. So I think they length of the reads are kind of strange. They range from 20-68 bp long. Does that seems a bit strange?
Would trimming the data to the same size help with the sequence length distrubtuion? or does it not matter that we have different size reads?
Would trimming maybe 7 bases from the start of the reads and maybe 3 bases at the end help with the per base sequencing content?
I just want to make sure we have our data trimmed like we should before we proceed. Do these flags matter for RNA-Seq data?
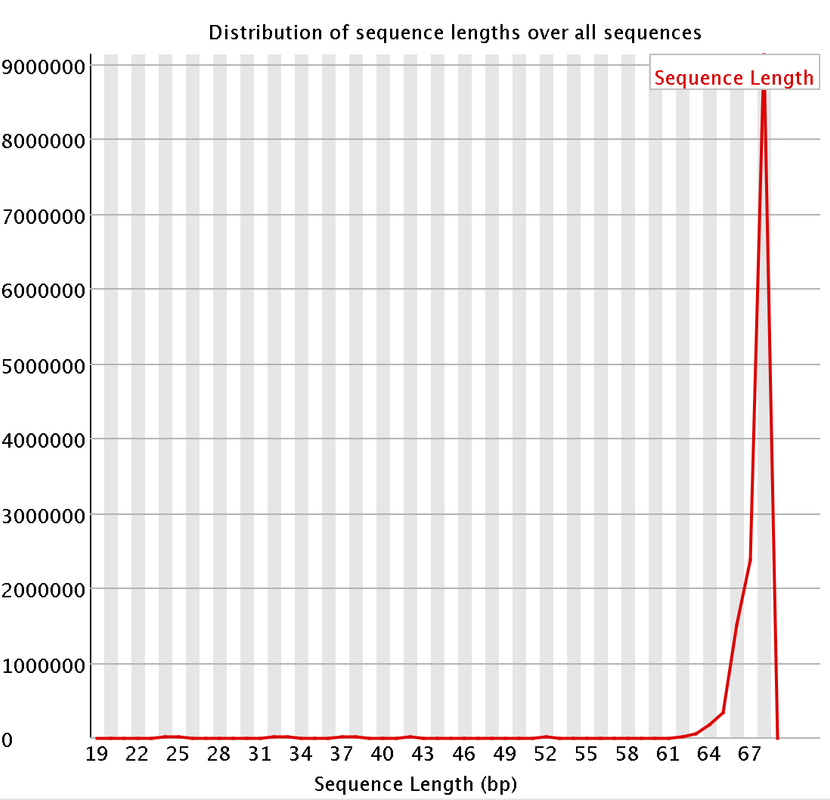
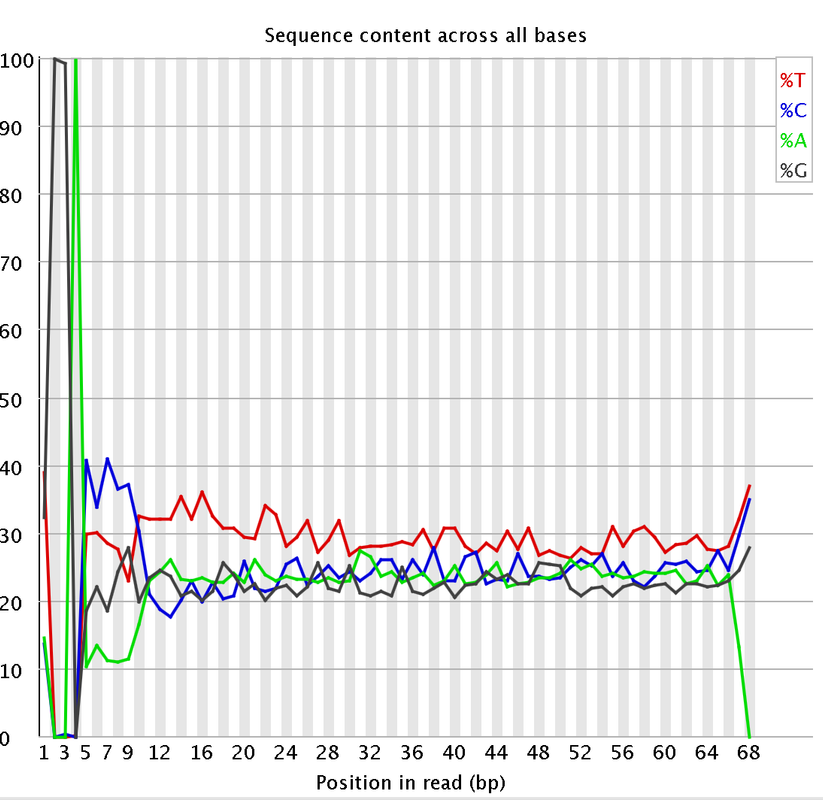


I did remove some universal illumina adaptors. Some of the samples had an crazy amount of universal adaptors. Something like 10% of the data. So you won't matter that the sequences range from 20-68 bp? Won't the 20 bp reads be more likely to map to the incorrect region?
Likely. If you are want to enforce a minimum length on the reads then you can filter them by using BBMap suite's
reformat.shlike this (replaceNNwith a number you want).If you have paired-end data then do
Do you have a suggested length? I think the majority of the reads will be around 64-67 bps long? Should I remove the remaining reads?
I just wanted to add, that like genomax said, although the distribution looks a little more extreme than usuall RNAseq data, it is characteristic for e.g. Illumina RNA-Seq data to show such bias in the first 12 nucleotides of the reads. This bias is generated during library generation and trimming the first few bases will therefor not eliminate it, since (as far as I know) it comes from site selection preferences during that process.
Thanks! I will keep those base pairs then!