Hi ! I'm currently a student and I have a hard time understanding some basics of bioinformatics; I'm currently learning about alignment, filtering, variant calling and such so my question might look silly but here it is anyway.
I have some trouble about how you work with paired-end sequencing files and what does it means to be paired-end.
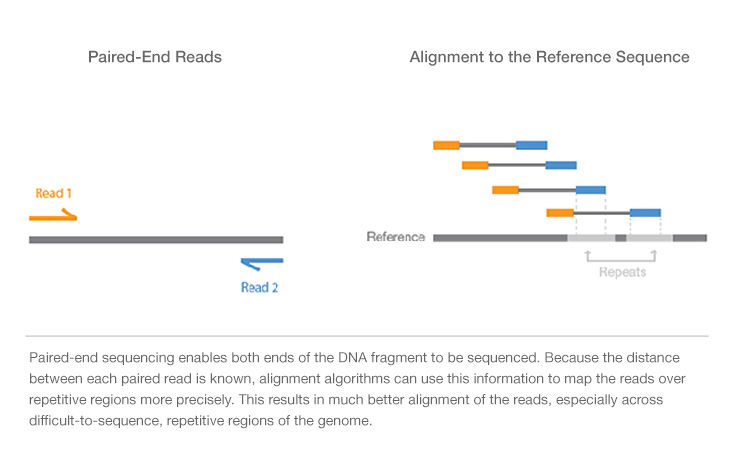
After taking a look on the Internet I found an explanation of what is paired-end sequencing (tell me if I got it right):
For what I understood, a paired-end sequencing is just done by sequencing from A to Z and then from Z to A. Which will provide two distinct datasets, one for each direction.
My question is, when you are doing some alignment with tools like BWA, TopHat or whatever, do you have to reverse one of the two dataset or not ? Because, for instance, If I wanted to find a consensus sequence (or the position specific score matrices), if half of the data are in the wrong direction wouldn't it be completely wrong ?
Completely unrelated: I've also heard that TopHat should be used over BWA for aligning RNA, do you know why ?