Entering edit mode

6.6 years ago
Morgan S.
▴
90
Hi,
I am working on fungal isolates from deep-sea sediments. It is debated on whether these isolates are haploid or diploid. I was wondering if there was a way to determine if they were either haploid or diploid using some type of bioinformatics tool.
I did try assembling the genome using both SPAdes and diploid SPAdes. Diploid SPAdes seems to be the better assembly, but I do not think this means I can ultimately say that these fungi are diploid.
Thanks in advance for your help!

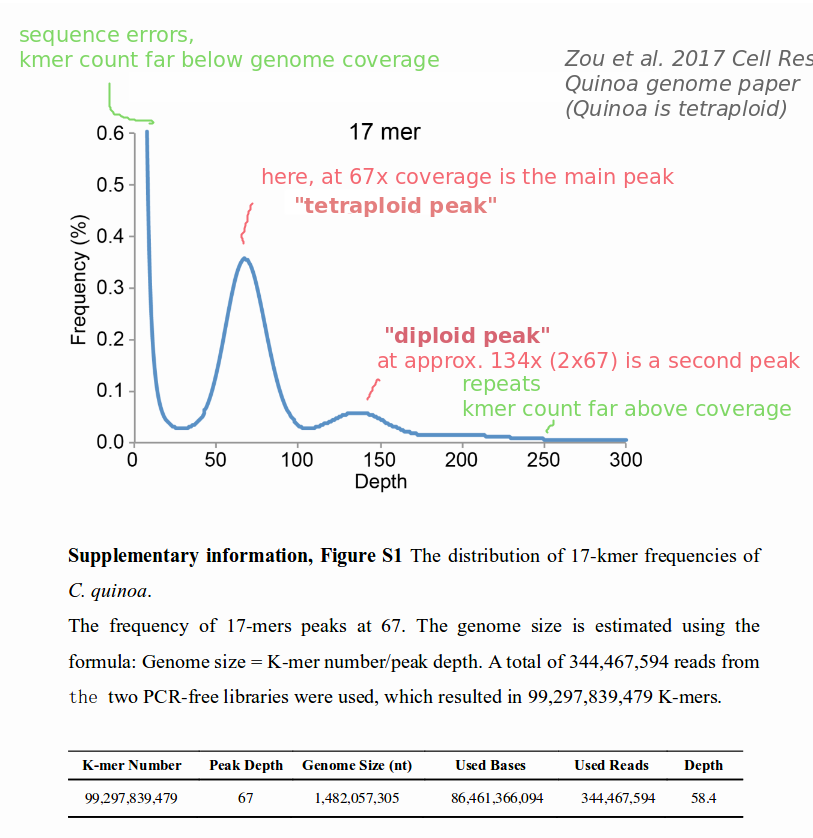

You can try
kmercountexact.shto test for ploidy (http://seqanswers.com/forums/showthread.php?t=64086 ).Additional link that is useful.