Entering edit mode

6.5 years ago
marongiu.luigi
▴
730
Hello,
I indexed the human grch38 assembly using BWA with the command
bwa index -a bwtsw <file>
I used a computer cluster and at the end, I obtained a use of ~75 Gb of memory. My PC has a memory of 64 Gb.
Does this mean that if I had indexed the file at home, the PC would have crashed? (BWA exits the computation when there is not enough memory allocated).
Is there a way to handle the memory usage so that the process can be carried out at home with the available resources?
Thank you
With 64Gb of RAM you can index 15 times the human genome using BWA
And btw a computer do not crash for this reason, if you do not have enought memory to run a program, it will just fail, the computer will be all good.
From here :
So why the cluster reported a total use of 75 GB? I previously tried with my PC at 48 GB but BWA stopped saying that it could not allocate several GB of memory...
which fasta file are you indexing? What's the name and where did you find it?
I got it from ftp://ftp.ensembl.org/pub/release-92/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna.toplevel.fa.gz
What is the size of your reference file ?
it is 1 GB zipped and 59.1 GB expanded
Something seems odd. Are you the only user on this machine with 64G RAM?
yes. actually, I did not installed the full 64GB yet, I still have 48 GB -- I am not sure if the investment of another 16 GB supplement would actually help. I launched the process again and the memory use increased to 29 GB (the gnome+chrome at rest consume about 3 GB, the rest was BWA) at 10 000 iterations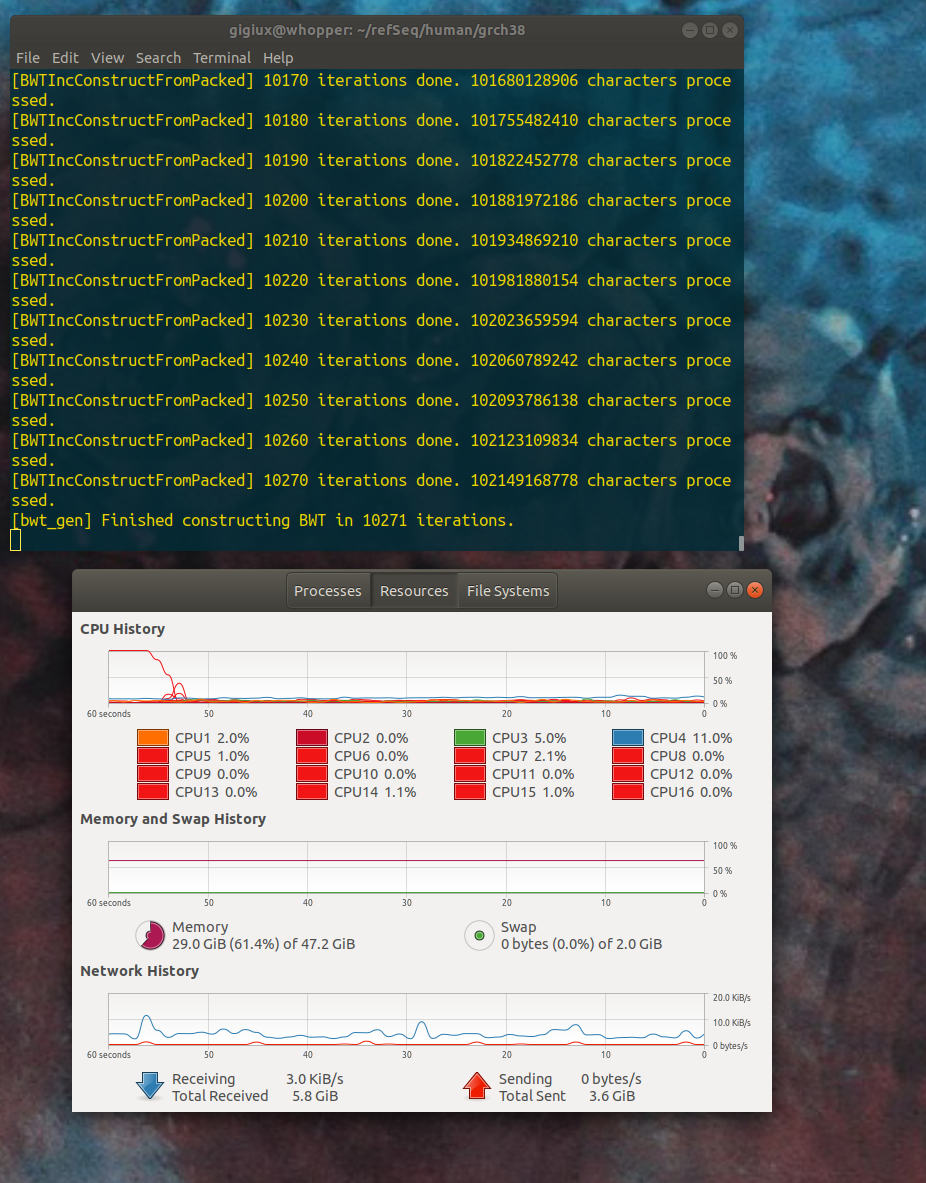 but the next phase crashed :
but the next phase crashed :
I interpret the error as the machine requires 51 GB of memory, that's why I wanted to reach 64 GB. But the multi-core computer used ~72 GB. for sure it's a lot above 5 GB.
I finally increased the computer's memory to 62.9 GiB, I launched
bwa index -bwtsw <file>but this time I got another type of error:What might have gone wrong this time?
I don't know how you manage your memory on your cluster but this error is still a memory exception error or a 0 division. Do you have more information about your error ? A complete log file ?
I am not managing it, I am simply running the same commands following the instructions from the manual; the rest is done by the operative system (Ubuntu in this case). How would I get the log file? those three lines were all that BWA printed out...
You are missing
-abeforebwtswin your indexing command. What happens if you dobwa index -a bwtsw your_file?good spot, I copied from the original post but I forgot -a over there. I re-edited the post.
Anyway, the outcome is the same:
and yet I now have available 6.7538E+10 bytes! What there is in btwtindex.c?
At line 158, that will be a command line to allocate memory in C language. I don't really know why your computer is gobbling your memory like this for a task like that...
marongiu.luigi : On a different note
bwaseems to accept gzipped sequence file. I am running an indexing operation with 50g of RAM to see what happens.It is odd since
hg38should not be that much bigger thanhg19in terms of size.You could try to pick up one chromosome from your input file and to index it, to see how your computer react with a smaller input file.
I can index smaller genomes with ease; I even indexed hg19. The problem arose with hg38.
BTW, a friend of mine also tried the indexing of GRCh38 to check, and also in his machine, BWA ate all the 40 something GB of RAM. So we have two independent operators and two completely different machines with the same issue.
It does not work even with
bwa index <file>:Could you give us a link to your file, the complete command you used and the bwa version please.
I already gave the location of the file: ftp://ftp.ensembl.org/pub/release-92/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna.toplevel.fa.gz. I have chosen this file based on the post remarking that Given the choice, use the Ensembl annotation. I renamed the file grch38.fa to short it up. The version of BWA is
Version: 0.7.17-r1188. The commands are eitherbwa index -a bwtsw grch38.faorbwa index grch38.fa.Did you unzip and rename or just rename?
See also http://lh3.github.io/2017/11/13/which-human-reference-genome-to-use
Unzipped and renamed. Dr Heng Li also sent me this link, I am trying this new version right now. I'll post the result...
I can confirm that the reference you shared above also caused my indexing to fail (16Gbyte RAM). The GRCh38 reference from the blog posted linked above is building fine - it seems.
Unbelievable, with the new file it was a piece of cake:
It took more or less half an hour and less than 10 GB RAM overall. This new file from NCBI has 6 184 133 946 characters instead of the 102 149 168 778 of the EMBL's. It is true that, nevertheless, they carry the same information?
Have you read the blog post of Heng Li? It explains what is included and what not. Alternatively, check for yourself which contigs/chromosomes are included in a reference fasta:
Yes I did, I just wanted a confirmation since seems incredible to me that 50 Gb of information could be actually shrunk to 3 Gb. Anyway, case closed: the indexing with BWA was finally done.
Further on this, is there also a VCF file associated with the NCBI reference genome? I tried with ftp://ftp.ncbi.nlm.nih.gov/pub/clinvar/vcf_GRCh38/clinvar.vcf.gz because it comes from the same source (NCBI), but I had problems with the headers while running GATK
A USER ERROR has occurred: Input files reference and reads have incompatible contigs: No overlapping contigs found.