Entering edit mode

6.4 years ago
salvatore.digiorgio
▴
10
I'm trying to develop pipeline to search for novel RNA editing events.
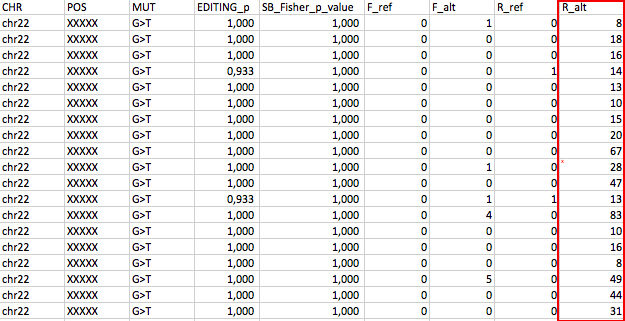
The last 4 columns are Forward_ref_cov, Forward_alt_cov, Reverse_ref_cov, Reverse_alt_cov
Could be artifact? How can i remove these kind of position??
Can you do this instead of the link above: How to add images to a Biostars post
Do you mean why the reads only align to the reverse strand and not the forward? Because RNA-seq or rather transcription is an orientation-specific event, and if a strand-aware library prep was used, then you see exactly what you see. If you need more details, please give more information on what you want to do.
Yes, I mean that some position i found show reads aligned only in one strand. I'm using TCGA RNA-seq data, and I read that unstranded library preparation was used, indeed most of my positions show mapping reads either in forward or in revers strand.
I moved your reply to comment in order to keep this thread organized. Can you provide a link to the exact source of the data?
The link of my data is https://portal.gdc.cancer.gov/. The information about data i used is at https://cancergenome.nih.gov/abouttcga/aboutdata/platformdesign RNASeqV2
and where does it say that this is an unstranded library? Could not find that right away following the link..
I'm sorry, the TCGA website is messy, and is difficult find the informations. This is what I found: http://embor.embopress.org/content/embor/early/2014/02/17/embr.201337950/DC26/embed/inline-supplementary-material-26.pdf?download=true
and on this blog there is just answered question at this link: A: Is TCGA PRAD RNA-seq data strand specific?
Did you do this mapping yourself? If so, you should provide details about how you did that.
Bam file I used are prepared according to this pipeline https://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/Expression_mRNA_Pipeline/ On bam file I used samtools and bcftools.