Hi All,
I have a question regarding trimming of adapters in NGS data. I have previously analyzed RNAseq data using brtim (https://www.sciencedirect.com/science/article/pii/S0888754311001339) without using adapter sequence. I am now analyzing smallRNAseq data, and I am using adapter sequence as adapter.fa using BBduk.sh from BBmap tools. Could you please clarify under what circumstances one would need to provide/know adapter sequence and when it is not necessary?
One case is where there is a kit/prep specific adapter that is being used. This may require special handling of the downstream data based on the instructions included in the kit.
If you had paired end reads with enough pairs having short inserts then you can detect them by doing this: bbmerge.sh in1=r1.fq in2=r2.fq outa=adapters.fa
This is a good question for the beginners who are starting with the NGS analysis.
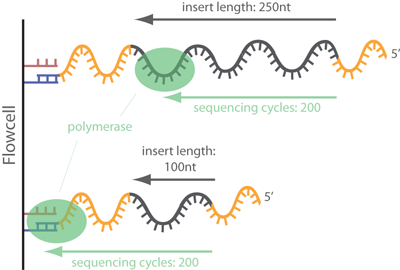
In common short read sequencing, the DNA insert (original molecule to
be sequenced) is downstream from the read primer, meaning that the 5'
adapters will not appear in the sequenced read. But, if the fragment
is shorter than the number of bases sequenced, one will sequence into
the 3' adapter. To make it clear: In Illumina sequencing, adapter
sequences will only occur at the 3' end of the read and only if the
DNA insert is shorter than the number of sequencing cycles (see
picture below)!
This is true in case of smallrna sequencing like microRNAs and piRNAs which are between 16-33 nt long. That means even with 50bp single end sequencing, 3' adapter sequences will occur. This is generally not the case with RNAseq where the gene/transcript fragments are way longer than the sequencing length and adapter sequences do not accur.
Also, if you are not sure which adapters are used for the sequencing, you can use an adapter predicting software like DNApi that will predict the most common adapter sequence which can be used by the tools like cutadapt or STAR before the mapping.
Paper: Tsuji, J., & Weng, Z. (2016). DNApi: a de novo adapter prediction algorithm for small RNA sequencing data. PloS one, 11(10), e0164228.
Thank you so much for this fantastic answer. I do have a few more questions related to this:
1. Reads in smallRna seq have adapters in 3 prime region only so, is it always right side trimming then?
2. Thus trimmed reads when aligned to the reference genome and saved as a bam file, why do we see both sense and antisense strands. This bam file shows both strands when viewed using IGV. Why? Shouldn't they all be 5' to 3' or unidirectionally aligned?
While the adapters may only be on 3'-end one may be asked to remove additional base pairs from 5'end. e.g. This NEXTflex smallRNA kit requires the following.
Clip the 3' adapter sequence (TGGAATTCTCGGGTGCCAAGG).
Trim the first and last 4 bases from the adapter-clipped reads
They also ask you
We do not recommend enabling the adapter trimming option or an adapter
sequencing into Illumina Experiment Manager, as it may lead to
unwanted masking of reads.
So always follow instructions included in kit (if one was used) or make yourself familiar with the read structure that would be expected.



Thank you so much for this fantastic answer. I do have a few more questions related to this: 1. Reads in smallRna seq have adapters in 3 prime region only so, is it always right side trimming then? 2. Thus trimmed reads when aligned to the reference genome and saved as a bam file, why do we see both sense and antisense strands. This bam file shows both strands when viewed using IGV. Why? Shouldn't they all be 5' to 3' or unidirectionally aligned?
While the adapters may only be on 3'-end one may be asked to remove additional base pairs from 5'end. e.g. This NEXTflex smallRNA kit requires the following.
They also ask you
So always follow instructions included in kit (if one was used) or make yourself familiar with the read structure that would be expected.
Thanks for explaining.