
In RNA-seq gene expression data analysis, we come across various expression units such as RPM, RPKM, FPKM and raw reads counts. Most of the times it's difficult to understand basic underlying methodology to calculate these units from mapped sequence data.
I have seen a lot of post of such normalization questions and their confusion among readers. Hence, I attempted here to explain these units in the much simpler way (avoided complex mathematical expressions).
Why different normalized expression units:
The expression units provide a digital measure of the abundance of transcripts. Normalized expression units are necessary to remove technical biases in sequenced data such as depth of sequencing (more sequencing depth produces more read count for gene expressed at same level) and gene length (differences in gene length generate unequal reads count for genes expressed at the same level; longer the gene more the read count).
Gene expression units and calculation:
1. RPM (Reads per million mapped reads)
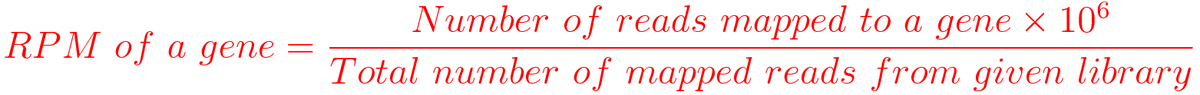
For example, You have sequenced one library with 5 million(M) reads. Among them, total 4 M matched to the genome sequence and 5000 reads matched to a given gene.
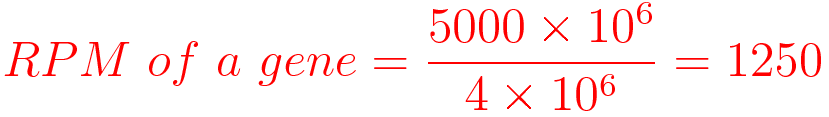
Notes:
- RPM does not consider the transcript length normalization.
- RPM Suitable for sequencing protocols where reads are generated irrespective of gene length
Here, 10^3 normalizes for gene length and 10^6 for sequencing depth factor.
FPKM (Fragments per kilo base per million mapped reads) is analogous to RPKM and used especially in paired-end RNA-seq experiments. In paired-end RNA-seq experiments, two (left and right) reads are sequenced from same DNA fragment. When we map paired-end data, both reads or only one read with high quality from a fragment can map to reference sequence. To avoid confusion or multiple counting, the fragments to which both or single read mapped is counted and represented for FPKM calculation.
For example, You have sequenced one library with 5 M reads. Among them, total 4 M matched to the genome sequence and 5000 reads matched to a given gene with a length of 2000 bp.

Notes:
- RPKM considers the gene length for normalization
- RPKM is suitable for sequencing protocols where reads sequencing depends on gene length
- Used in single-end RNA-seq experiments (FPKM for paired-end RNA-seq data)
3. TPM (Transcript per million)
Notes:
- TPM considers the gene length for normalization
- TPM proposed as an alternative to RPKM due to inaccuracy in RPKM measurement (Wagner et al., 2012)
- TPM is suitable for sequencing protocols where reads sequencing depends on gene length
Detailed article: https://www.reneshbedre.com/blog/expression_units.html
References:
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature methods. 2008 Jul 1;5(7):621-8.
- Wagner GP, Kin K, Lynch VJ. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory in biosciences. 2012 Dec 1;131(4):281-5.

The M (per million) is for normalizing for sequencing depth.
The difference between FPKM/RPKM and TPM is the order of operations.
It should be stated up front that neither of these methods is optimal for conducting differential expression analysis across samples.
what about rpm/bp?reads per million mapped reads per base pair (rpm/bp) with background subtraction