
I used pruned SNPs from 1000G phase 3 to do a PCA, and got the following results:
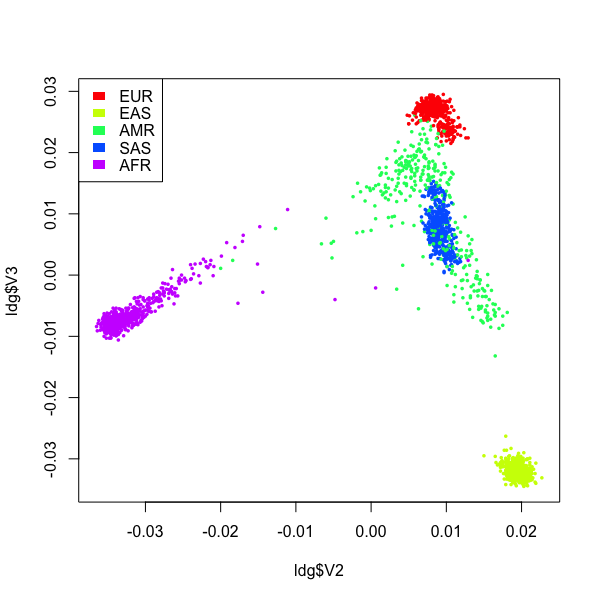 In the plot above, different populations were well seperated, and individuals from the same population clustered tightly. ~ 20,000 SNP were used.
In the plot above, different populations were well seperated, and individuals from the same population clustered tightly. ~ 20,000 SNP were used.
I then merge 1000G data with data from my study population, and PCA results looked like:
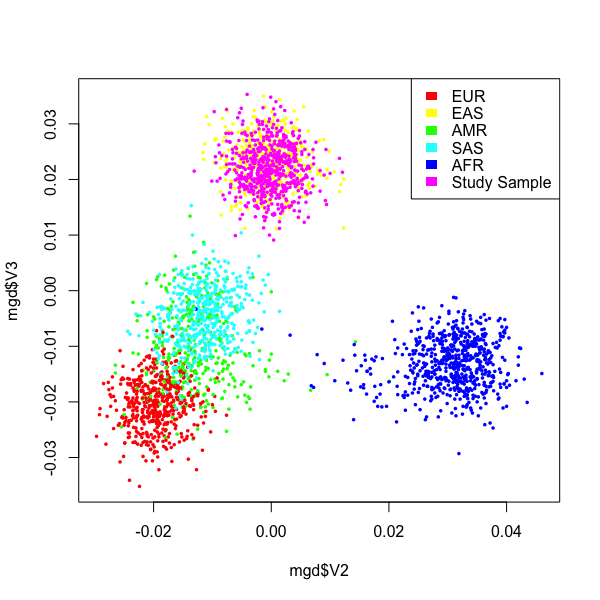 This figure is generally right. But, the points are quite scattered, and individuals from different ancestries is overlaping. I think this is because the number of SNPs used in PCA is not sufficient. Indeed, I only have ~ 900 SNPs after merging my data set with 1000G SNPs. Notably, my data also has about 20,000 SNPs before merging.
This figure is generally right. But, the points are quite scattered, and individuals from different ancestries is overlaping. I think this is because the number of SNPs used in PCA is not sufficient. Indeed, I only have ~ 900 SNPs after merging my data set with 1000G SNPs. Notably, my data also has about 20,000 SNPs before merging.
So I am wondering why there is so few SNPs after merging 2 datasets, and how to deal with it?
Thanks!
How did you merge the datasets? Please be as complete as possible when asking questions and include commands used.
I merged it with mergeit to in EIGENSTRAT. The tool merge two sets by finding the union of individuals and intersect of SNPs.
Is your data from a microarray?; if 'yes', then which one? Did you perform any LD pruning?
Actually, I have WGS data. I read a Nature Protocols article on GWAS QC and realized that I should use the complete SNPs from reference panel (1000G) to merge with my data first, and then extract SNPs from LD pruning that was done in my sample. However, I did LD pruning for both 1000G and my own data set, and this might be why there were so few SNPs after merging. Thanks!
Yes, the part where LD pruning is performed is key, i.e., before or after merge. Best of luck!