
Hi all,
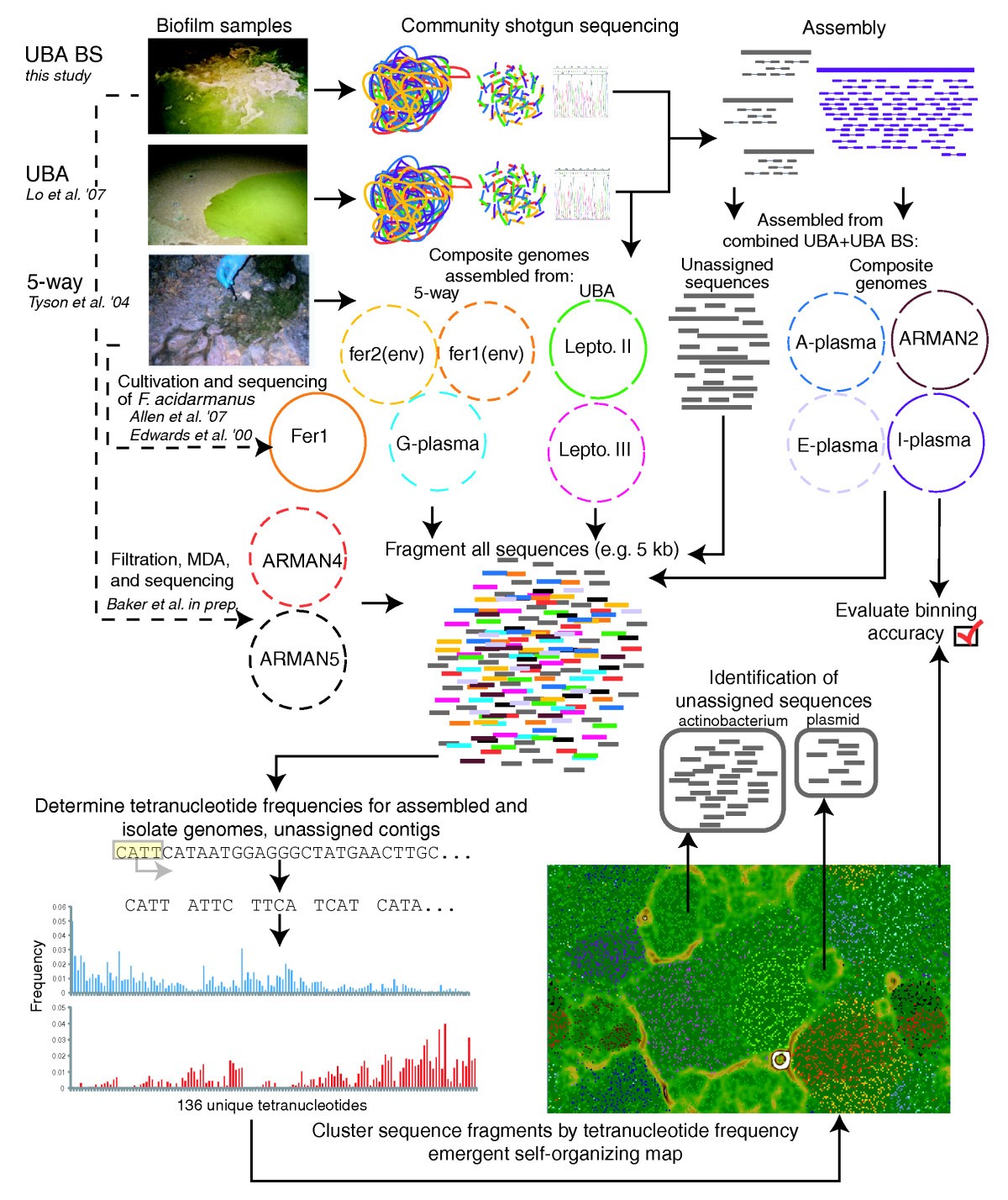
I have a couple of metagenomic samples in the form of genomic assemblies and I wanted to perform binning and generate ESOM (Emergent self organizing maps) similar to the one mentioned in Figure#1 of this paper. The primary objective is to generate ESOMs. I have tried to go through some papers and I found that binning could be performed based on genomic signatures (i.e. di, tri or tetra nucleotide frequency) generated from metagenomic contigs. There are infact few scripts available to generate those signatures ; for e.g. this one here which could be used as input to databionics ESOM tools.
I have a couple of questions:
Apart from the metagenomic contigs, is there anything else that is required as input or to generate necessary files to be used as input to ESOM tools?
The installation page for ESOM tools mentions to download some files required for MATLAB. is MATLAB absolutely necessary to generate ESOMs? MATLAB is a paid software as per my information.
Finally, can someone point me to a good tutorial/ material for a step-by-step guide to generate ESOMs?
Let me know if someone requires additional details to answer my questions.
Thanks Vijay

{kind=link}
Dear 5heikki
Thank you for the response. As I mentioned, the ultimate goal is to generate ESOMs ( that's the requirement of the project ). Maxbin, though will be easier as you mentioned, I think , will not provide emergent maps ; correct ?
Additionally, I am looking at "Calculation of tetranucleotide frequencies and clustering by ESOM" section of the paper. Will this script help me generate tetranucleotide frequencies ? Have you tried it ?
The U-matrix or whatever presentations of the ESOMs themselves don't really serve any purpose apart from looking cool. You're correct about MaxBin not producing ESOMs. However, the ultimate goal, i.e. binning, is the same. I don't remember exactly what I did as it was many years ago. That script at least says that it will generate those frequencies. Do you have any reason to doubt that it won't?
Hi 5heikki
Thanks for the input once again. So, I was able to follow the instructions on the gihub page and thankfully all the programs worked as expected. For the sake of quick start, I just tried this with 2 files
file 1 is the e.coli genome and file 2 is a metagenomic assembly. Now, after the training finished (which took 19 hours to complete - posted that on their github page as an issue) , I have this image:
However, I want to produce an image something like this,
SOURCE: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4177395/figure/F1/
i.e. making it much more informative by adding taxonomic information. What should I do ? Any further inputs ?
AFAIR you can manually select and color regions with the Databionics GUI. The rest of that was probably made with PS or some other image editing software..
I am not sure whether this question is still relevant to you, but maybe it might help others experiencing similar issues:
You could circle your data points according to the topography of the map and save the resulting *.cls file. Then, you can combine your windows/contigs into seperate files depending on what bin they were grouped. You could blast these fasta-files against genbank and see whether you can see significant hits.
If you can actually connect some of the bins according to the species, you can either rename your classes or number them as in the picture you entered here, and then manually add the taxonomic name below (in addition to the color and number).