Entering edit mode

6.6 years ago
dcheng1
•
0
For ChIP-seq data of H3K4me3 histone modification. I used MACS2 to call peaks with default setting. However I identified a large number of peaks with length below 700bp. Those peaks look like vertical bars in IGV. Any opinions are greatly appreciated!!
Below is a subset of the narrowPeak file:
chr10 131364552 131365161 Peak_1 127 . 7.76171 12.79630 3.30580 286
chr8 133508303 133508979 Peak_2 93 . 6.03689 9.34294 2.21605 337
chr12 49445522 49445922 Peak_3 87 . 5.86739 8.77625 1.76287 219
chr10 73439426 73440041 Peak_4 76 . 5.17447 7.69945 0.80224 295
chr11 114011885 114012344 Peak_5 76 . 5.17447 7.69945 0.80224 177
chr11 1213144 1213654 Peak_6 76 . 5.17447 7.69945 0.80224 225
chr1 11661364 11661760 Peak_7 76 . 5.17447 7.69945 0.80224 144
chr11 66866515 66867011 Peak_8 76 . 5.17447 7.69945 0.80224 344
chr11 77008922 77009283 Peak_9 76 . 5.17447 7.69945 0.80224 242
chr1 201072066 201072625 Peak_10 76 . 5.17447 7.69945 0.80224 334



So what's your question? H3K4me3 peaks are often relatively narrow.
Yes, H3K4me3 peaks are narrow. However I usually observed peaks with average length 2kb in high quality data. The short peaks(~700bp)look like spikes, not bell curved shapes.
maybe sharing a snapshot of such peaks will help us better understand any issues you may have with their appearance
How did you make your bigWig file? The 'fc' in the file name makes me think those values are fold change values for each region rather than actual reads. What does the BAM file look like for the same region if you throw it in IGV?
I think you're correct, the signal track is for fold change. I used AQUAS chip-seq pipeline to get this bigwig file.(https://github.com/kundajelab/chipseq_pipeline)
Below is the snapshot for the BAM file and fc bw file opened in the IGV: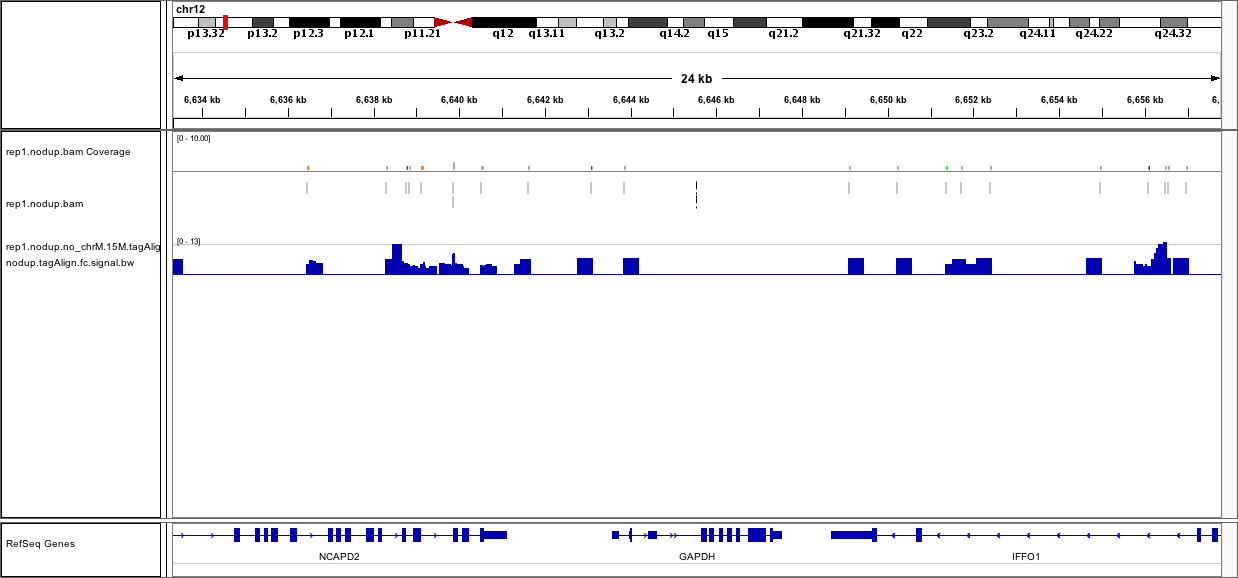
I just realized for the above shown data, the reads is only 2 million. Below is another data with 6 million reads: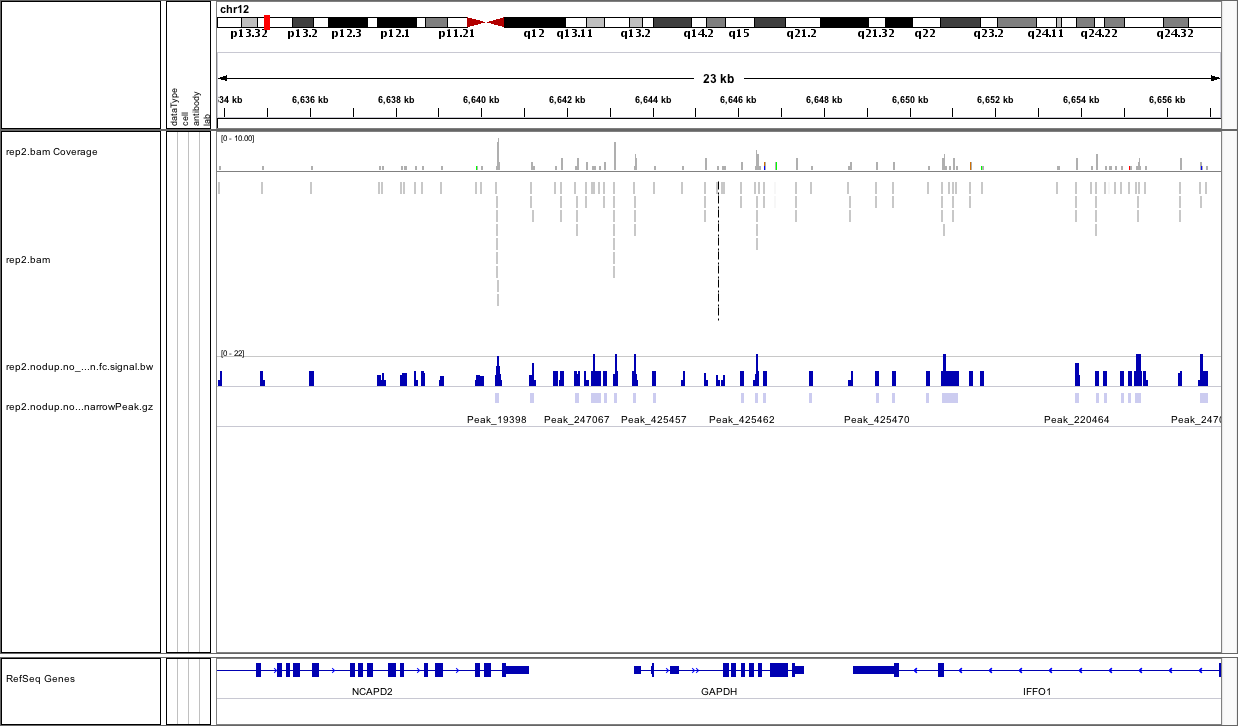
This data quality isn't looking too great to me. Seems like the coverage is just really low and that perhaps the ChIP didn't work very well - the GAPDH promoter should be a mountain in most decent quality data sets. I'd be skeptical of this data.
looks like fairly old (mod)ENCODE data...certainly not ideal and the problem is probably not primarily with the peak calling but the coverage as Jared pointed out