Entering edit mode

6.2 years ago
ductylerlv
▴
10
Hello All,
Our lab is getting some interesting results whereby we have modules that have both positive and negative kme values in a signed network. If a higher power is used this goes away but the lower power already surpasses the R2 and k.means thresholds. Any insight would be helpful.
Best,
Duc

can you post the code ?what you had done?
I was the one who encountered negative and non-correlated kME values for specific network nodes in WGCNA with the following input and code. The soft threshold criterion for scale free topology was met at power, beta, 7.6. We chose 8.5 to reduce noise further.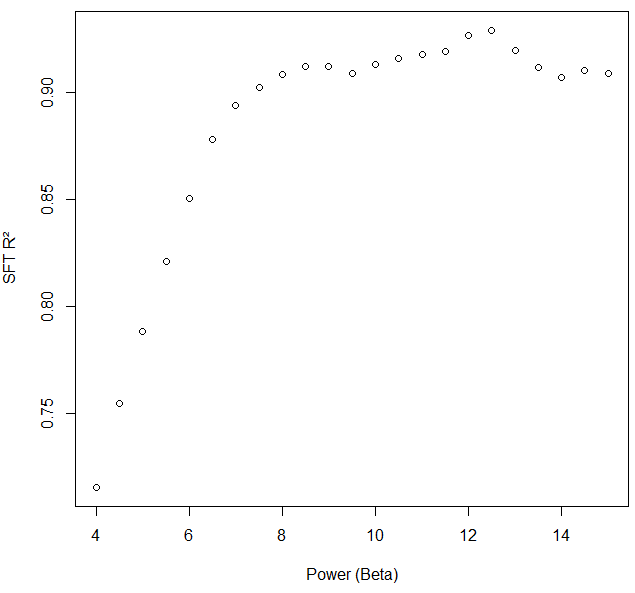
And the resulting protein network has module(s) like the below violet module, where we never usually see kMEintramodule<0.20, but here we do: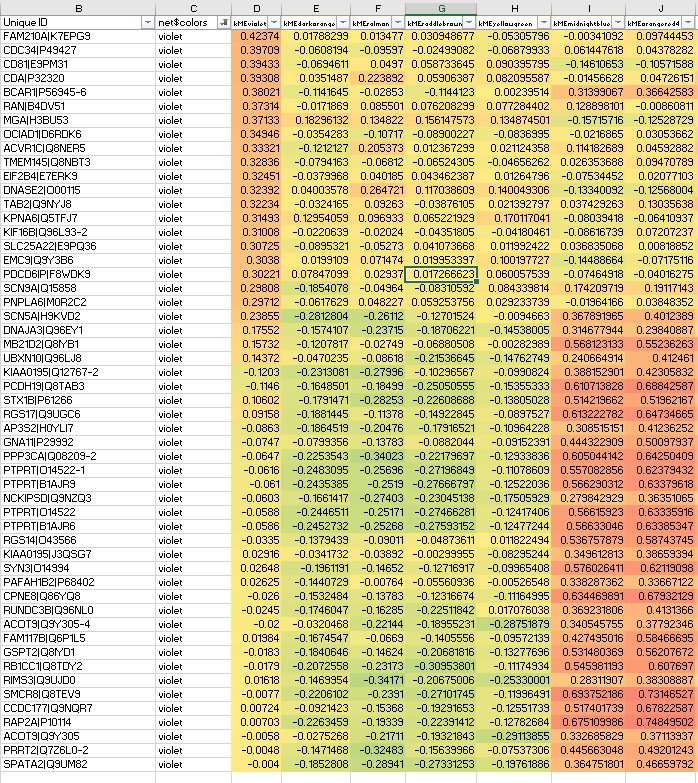
Clearly some of the lower violet module members should go to orangered4 with kMEorangered4>0.7.
Interestingly, some orangered non-hub members suffer from anticorrelation to their own ME, precisely what should not be seen in signed network modules: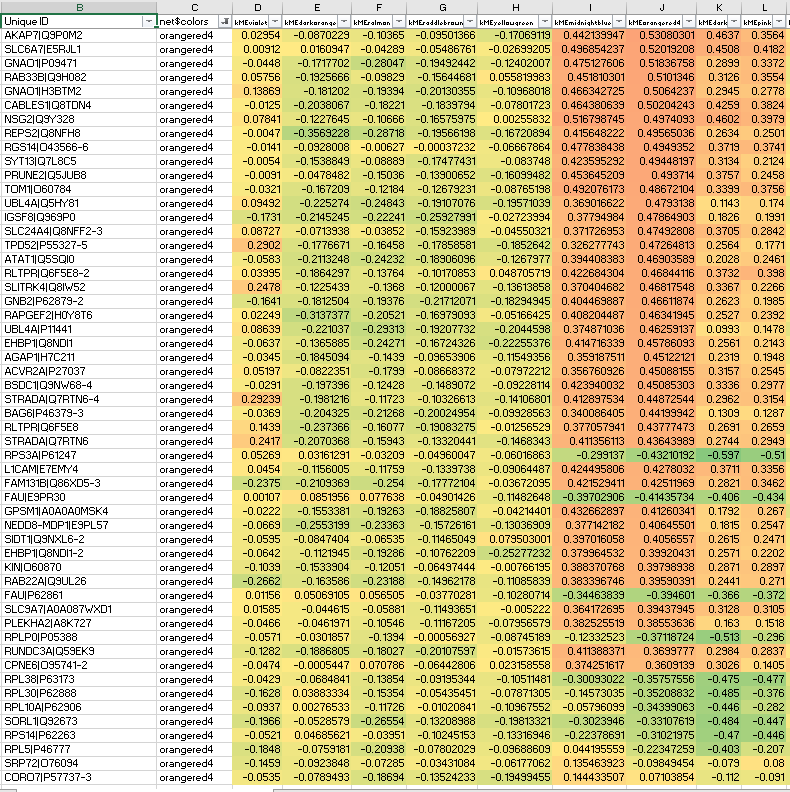
Full code to reproduce the problem follows:
Formatted ModuleAssignments (kME table) can be found here: Excel Binary kME Table for above network output
Input for cleanDat log2(abundance) matrix loaded in above code is here: ZIP with log2(abundance) matrix text tab-separated file
...explicit use of minKMEtoStay=0.30 has never been necessary before.
I should add that minKMEtoStay=0.30 is a default parameter/setting: blockwiseModules in WGCNA v1.64 default parameters
Rerunning the above blockwiseModules() function with this parameter added to the others gives identical modules.
My sessionInfo follows:
Did I provide too much information? Hoping to get some feedback.
Perhaps this answer from the WGCNA developer helps? - https://support.bioconductor.org/p/101579/