
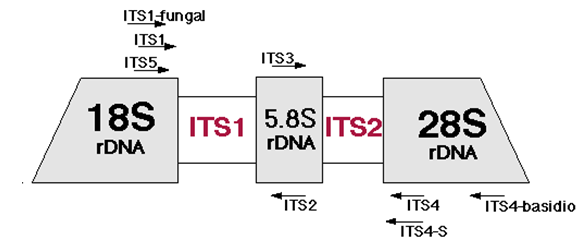
I would like to use my lab's MiSeq to seqeunce the ITS region (which include ITS1, the 5.8S gene and ITS2). For the species of fungus I would like to look at, this sequence falls within the ~750 bp range.
I have read several papers and known labs to use the ITS5 and ITS4 primers to capture this entire region, they would then use Sanger sequencing to capture this ampllicon region. Obviously 750 bp is too large of an amplicon for a MiSeq to capture in one read.
A method I would like to try is using ITS5 and ITS2 to capture half of the region and ITS3 and ITS4 to capture the other half. I would then like to recombine these reads and then align them to an ITS region database.
If anyone knows a tool that would be helpful to combine the amplicon sequence halves back together, please let me know.
Thank you in advance!
Not what you are asking for, but I expect the "less messier" way to just align those 2 amplicons separately, looking for genomes in your database to which both map...
Thank you, this does sound like a cleaner and more practical approach.