Entering edit mode

6.2 years ago
kathrine.tan
▴
10
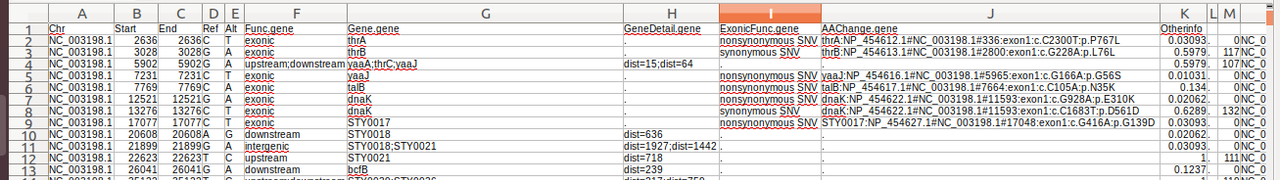
I am new to the annotation of variants using annoVar. I try to understand the values listed in the column "Otherinfo" (Columns : K & M) by reading the documentation of annoVar. However I am still not clear about how these values were generated.

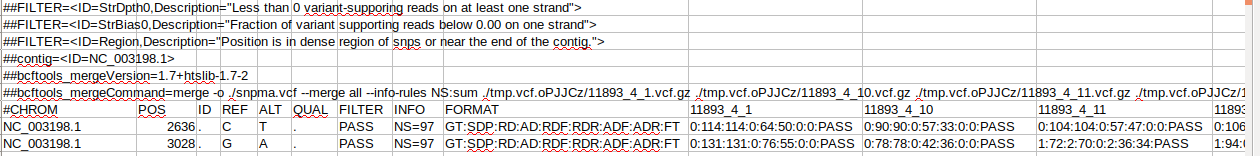
The column K looks like SIFT score. This information come from the database you add in you command line for the annotation.
Dear Titus,
Thanks for answering. I will try to look at it.
K