Entering edit mode

6.0 years ago
Morgan S.
▴
90
Hi,
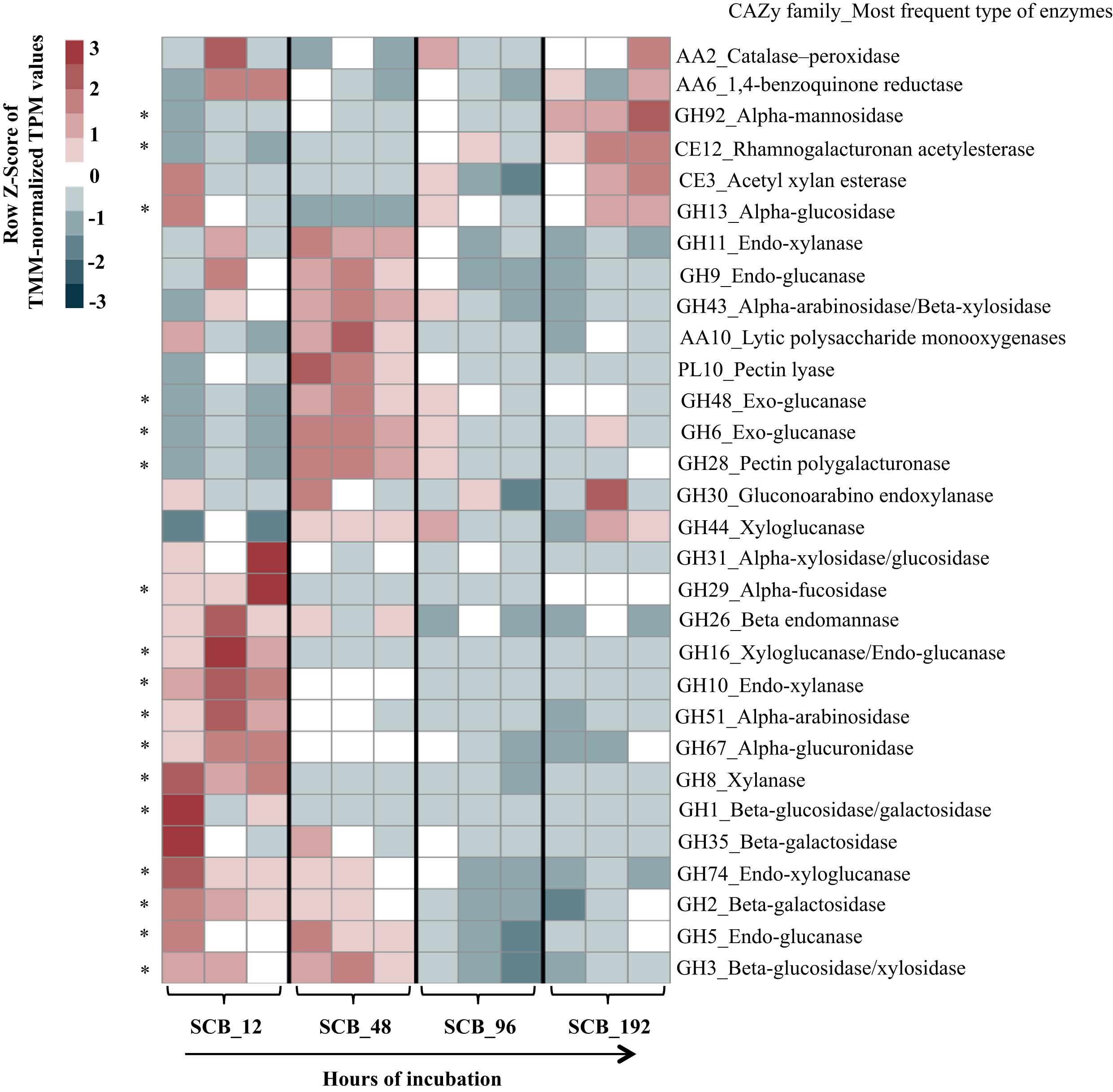
I am annotating two fungal genomes. I have run the genomes through dbCAN database to determine how many of the putative proteins encode CAZy enzymes, but now I am trying to figure out if I can simply report the relative abundance of the genes for each genome, or if I need to show some sort of normalization of the number of enzymes?
Many papers that I have read normalize their reads in some way, but I believe that this is because these are based off transcripts, not a "near" complete genome.
Like shown here in figure 2:
{kind=link}
I hope this makes sense. Please share your thoughts.
Thanks in advance! Morgan