Entering edit mode

6.0 years ago
Pin.Bioinf
▴
340
Hello,
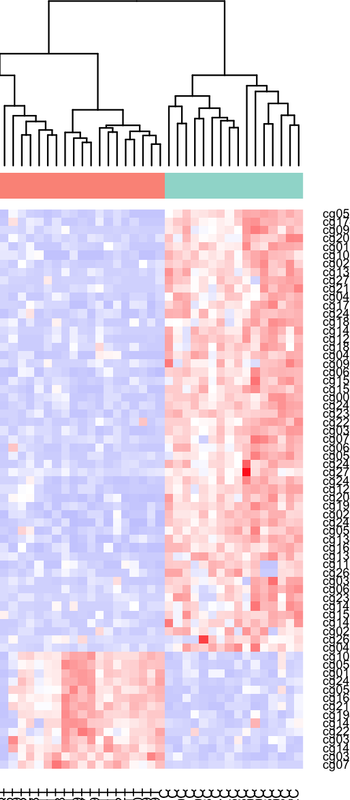
I have plotted my methylation beta-values of 1000 CpGs in tumor-controls. The heatmap seems ok at first glance, but I see that some CpGs that are differentially hypomethylated in controls appear as hypermethylated in controls in the heatmap. Is this because of the z-score scaling? Should I convert to M values first? Thank you.
EDIT: Here is some code:
Matrix (only an example of the big matrix):
SAmple1C Sample2T Sample3C Sample4T Sample5C Sample6T
cg03185752 0.03128 0.06667 0.00079 0.13409 0.01614 0.00000
cg26222311 0.10023 0.00000 0.08416 0.00000 0.04864 0.00000
cg13488570 0.30099 0.06393 0.29301 0.02862 0.39648 0.00343
Code:
heatmap.2(Matrix,scale='row',col=bluered(149),trace='none',
main = "Centered CpG sites",ColSideColors=rep(c(col1[1],col1[4]),19),margins = c(9, 22),density.inf="none",symkey=TRUE)
legend("topright", title = "Samples",legend=c("Control","Tumor"),
fill=c(col1[1],col1[4]), cex=0.8, box.lty=0)
Result:
Share some code, so we can see what you're doing.
I just edited thanks.
The
heatmap.2function usesscale = "none"as default. If you find hypermethylated values, as hypo in your heatmap, you probably made a error before the z-score conversion. Maybe switched control with treatment group somewhere?What I expect to see is all hypomethylated tumors, no hypermethylated tumors, which makes sense in most of the heatmap, but at the bottom there is an inverse behaviour : hypermethylated tumors and hypomethylated controls. the DMPs calculation results in those CpGs show hypomethylation in tumor, so the heatmap should reflect the same thing in those cpgs right? or are z scores messing with it?
I don't see any code making z-scores, so I cannot judge if they are messing with it or not...
I thought scale='row' did the transformation to z-scores? that is all i am doing, introducing a beata value matrix into heatmap.2 function and setting scale to 'row'
Right, I overlooked
scale='row'in your code, that's why I was talking about default setting. Try to make your own z-scores first, and than make aheatmapwithscale='none', see if it looks better.