
Hi there, We sent in a ribosomal and total extracted RNA to the company Novogene for analysis and something strange has happened with the data. A key marker gene (almost essential to the analysis) is missing reads. Every single publication concerning JA has had reads for this wound induced protein (okay, didn't check all, but I've made sure many have). It's not even a question. It's there. It's there in decently high quantities. It's a published fact. My postdoc has additionally confirmed the presence of this gene using qPCR. It's not a part of any hypothesis. It's a fact that it is induced at this point.
However, out of the whole genome, all the biosynthetic genes for JA, all the important high and low induced transcripts, this one gene is said to have 0 reads.
What gives? The bioinformatician has been very unhelpful - he/she seems nice, but hasn't been reading the emails properly and answering the wrong questions. His latest reason is that every single read (there are lots seemingly aligned to the genome (IGV viewer and bam files, TAIR10), especially when compared to other genes that have reads) fell into the 3 out of the 8 categories for being unable to have the read be quantified (HTseq - 5 out the 8 circumstances can be quantified). This gene doesn't have an overlapping genes or an intron.
Is this actually possible? That a published, verified, marker gene seriously can have no reads whatsoever? And in the same RNAseq analysis transcripts that are expected to have little to no reads can be quantified? We also have all the data showing the quality control was passed and the vast majority, almost all, reads mapped.
Sorry for the harsh tone, it's been a frustrating few weeks.
Did you make the libraries or did Novogene do them? If you made the libraries and used a stranded kit/protocol then that information may not have been passed on to the analyst. Perhaps this is the simplest explanation. Once the strandedness is taken into account everything should get sorted out.
An off-line discussion with @Ian and @Devon led to additional points that you can consider :
If you (or Novogene) did use a non-stranded prep then you may have to do qPCR to determine the level of the gene on the other strand and compare it to qPCR values for the gene you are interested in to determine the ratios.
Using Salmon will likely allow you to assign proportional counts to the two overlapping genes.
I can't thank you enough for all the help and advice with this issue. Novogene did the library-prep. Does this mean our whole analysis may be compromised if they used the wrong setting/parameter in HTseq? We've done the qPCR for MYC2 - and we have positive results - expected results. We have not done pPCR for the oppositely stranded gene.
I will try Salmon! I'm excited we have a real reason for this to have happened.
While I don't have first hand experience @Ian.Sudbery mentioned that the default prep Novogene does is un-stranded (NEBNext Ultra). That is what they may have used in your case, if you had not specifically asked for a stranded prep option (check with them for specifics).
In that case the strand information is lost when the libraries are made so you can't be sure if the expressed sequence came from one gene or the other (since they are overlapping). You know by qPCR that your gene of interest was expressed but you would need to check if the other one is or not. If the other one is not expressed at all then counts you see would be for your gene of interest.
Oh wow. That's concerning..the read results for the other gene are not good (quite frankly, they suck - the fpkms are like 0.6-0.9). The homologs of MYC2 (MYC3, MYC4, MYC5, MYC1) have better reads (fpkm of around 30) and they are not important, but they have actual reads!
I'm so worried our whole experiment is compromised now. I'm about to email the bioinformatician with all these inquiries in a well formatted manner that's not too long so he will read it - hopefully.
If he says anything contrary to you/everyone here, I'll respond back. Thanks again for all the help!
Hate to butt in but FPKM is an old unit of measuring mRNA expression that was defined when RNA-seq studies had very small sample n of, like, n=1. With no cross-sample normalisation when deriving FPKM units, values are actually incomparable across samples and unsuitable for differential expression comparisons, at least, but should be roughly comparable within sample. Probably just something to note for future reference.
FPKM <1 seems like noise to me, to be honest. 30 is not high but would indicate expression.
Please see @Kevin's answer : calculate p-value/q-value use fpkm - RNA-Seq
Well, you are certainly not the first person that I have come across that has expressed some frustration at a service provider for a bioinformatics analysis. However, without knowing each and every step that was performed by the company, it is difficult to say what went wrong. As you indicated that HTseq was used, however, my gut feeling is that it relates to reads that 'multi-map' - see the second question in the FAQ, here: https://htseq.readthedocs.io/en/master/count.html
Knowing how the reads were aligned in the first place is important, too. Did they choose the correct alignment program with correct parameter configuration? - you could probably ask.
So, it is a single exon gene / non-coding RNA?
HTseq would not be my choice at all for RNA-seq, by the way. I would use Salmon, Kallisto, or STAR+featureCounts for the purposes of read count quantification.
I will see if I can get others to respond.
I have tried what Kevin suggest a couple of times.
Hi! It is a single exon gene. I was corrected and in the "Ensembl_release_40 of Arabidopsis thaliana there is overlap between At1g32630.2 from 11797937 to 11798676 At1g32640.1 from 11798119 to 11801407"
However - how does that explain overlap? We just received that exact text in an email today from the bioinformatician. I'll upload the screen shot.
Is that built (the ensemble_release_40) from or the same as the TAIR10 genome? I'm actually lost with the difference there.
I ask that because my professor explicitly asked for TAIR10 to be used and the IGV viewer is aligning the reads to TAIR10. I hope that's not a dumb question.
That's TAIR10, it's just an updated annotation where the 3' end of both genes have been extended (there's going to be evidence for why) so it overlaps with At1g32630 heavily. Note, however, that they're on different strands, so unless you used an unstranded library preparation protocol this won't be an issue (use the appropriate strandedness setting in featureCounts or htseq-count).
With the amount of information available to us, I think Devon Ryan explanation above is the best guess you will get. To know if the lack of counts is due to an bioinformatics analysis error or due to limitations in the methods used, we would need to know if a stranded or unstranded library preparation was used, the exact commands used to perform the bioinformatics analysis, and the annotation version used for read quantification.
To add a visual aid to Devon Ryan explanation, here are two screenshots from Ensembl and NCBI genome browsers:
Ensembl:
NCBI:
Genome and annotation versions are confusing in plants. In vertebrates, the genome version (that is the DNA sequence of the genome and its arrangement in contigs) and the gene annotation version (that is what the sequence of each transcript is and where it is transcribed from the genome) are different things (although a particular gene annotation will be based on a particular genome version). So for example hg38 is the current version of the human genome, and Ensembl version 93 is a gene annotation on that version of the genome.
In plants this is a bit confused. TAIR10 is a genome version. But when TAIR10 was released, it was released with a gene annotation, also referred to as TAIR10. However, there is a more recent and comprehensive gene annotation called araport11 (still based on the TAIR10 genome sequence). Araport11 is NCBI GenBank and Ensembl's preferred Arabidopsis annotation and Release 40 of Ensembl plants is based on it. Even the TAIR website displays araport11. However, it only displays what it has decided is the representative transcript of each gene.
The old "TAIR10 release annotation" is still available on the TAIR website, but this is not what I would recommend. See @Deven Ryans comment below - find out if your sequencing was stranded, and if it is htseq-count (or feature counts) can be run in the appropriate stranded mode. Alternatively use on of the quantifiers that can cope with such ambiguity (salmon/kallisto/RSEM) or ask the bioinformatician to do the quantification over the protein coding part of the transcript only.
That is a little confusing. Thank you for your extremely helpful explanation!
Have you/your post-doc looked at the alignments yourself using IGV (it is not clear from the post above though the title seems to indicate that someone has)? This requires no special bioinformatics expertise and anyone can do this on a PC/Mac (only Java needed). I assume you have been provided the aligned data files. If not, you should ask that co-ordinate sorted/indexed alignment files be provided.
If no reads actually show up in IGV as aligning to this gene (that you expect to be there) then there is something wrong with either the analysis, sequencing, libraries or your samples (which appear to have been validated by qPCR, so perhaps they are fine). You will need to trace the problem back up that chain.
Can you clarify what this means? As @Kevin said above, if a read is multi-mapping, then that is the only time it is not going to be counted (by default).
I believe that the novogene bods will have been referring to this from the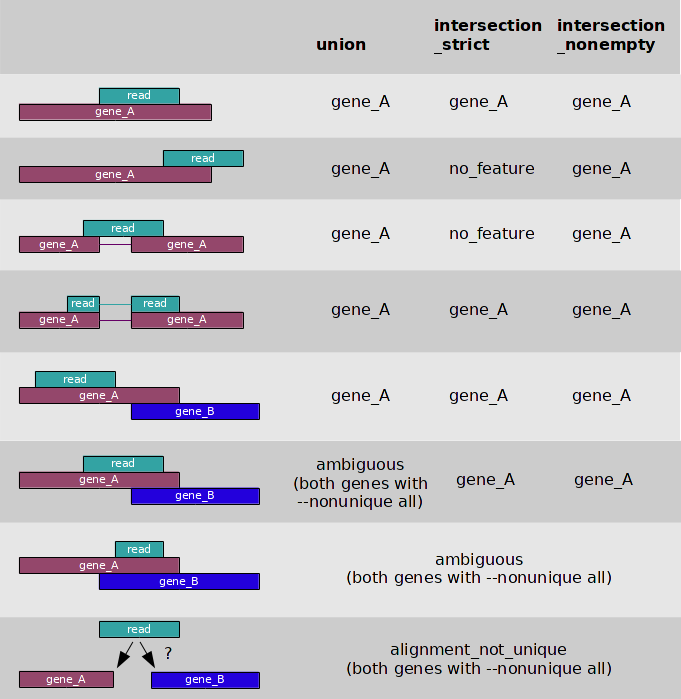
htseq-countmanual:Given that they are referring to 5/8 circumstances, my guess is that they are using the union column, and you say there is no overlapping genes (are you sure this is true? Not even weird none-coding transcripts?), I'm going to guess the problem is multi-mapping reads.
Its fairly uncommon for every position in a gene to be non-unique, but not completely unheard of. One thing to be aware of is that I have seen bioinformatics service providers attached to sequencing companies use non-spliced aligners for aligning RNA-seq data - I've even seen the Broad do this! In general it doesn't work as poorly as you might imagine, but if you have long reads on a short gene with an intron in the middle and this is the only unique sequence, you might have a problem.
There is nothing really wrong with HTseq-count, compared to featureCounts, but neither can deal with multimapping reads. If this is the case, then I second the recommendation to use Salmon/Kalisto/RSEM with this data as they deal gracefully with multi-mapping reads. Both Salmon and Kallisto are easy to use and do not require particularly high powered computers. We've successfully used Salmon on Arabidopsis data using TAIR10 annotations may times, including looking at the JA synthesis pathway.
Thank you so much. Yes that is the exact diagram he sent us to help explain what could be going wrong. They have done other analysis (RNAseq) for other labs and those labs have published read counts for MYC2. How could they get around the overlap for that labs RNA sample, but not ours? Does that make any sense?
Additionally, the other gene has has reads the At1g32630. Why did all of MYC2s get thrown out?
@rbkh09, can you tell us which gene exactly you are reffering to?
The gene is a transcription factor: Myc2 or At1g32640