
Hi all,
I am new to bioinformatics and am working on a school ML project that seeks to use the DNA Methylation data extracted from TCGA, to identify cancerous tissues from normal/control tissues, as well as distinguish between the different types of cancer (i.e. BRCA v.s COAD).
I have downloaded the "Illumina Human Methylation 450" level 3 data-set and will like to know how can I tell if a sample is cancerous, or normal/healthy since it is not indicated and I will require the information, in order to create a training data set.
The headers included in the data-sets which I have downloaded are as follows:
1) Composite Element REF 2) Beta_Value 3) Chromosome 4) Start 5) End 6) Gene Symbol 7) Gene Type 8) Transcript ID 9) Position to TSS 10) CGI Coordinate 11) Feature Type
This seems to be quite different from the headers present for another similar study (https://arxiv.org/ftp/arxiv/papers/1804/1804.04839.pdf) as they have a header which states the class of the tumor (normal or cancerous).
Thus, can I check if the "class" header is present on level 2 or level 1 data instead? Or is it distinguished via the assigned TCGA barcode number:
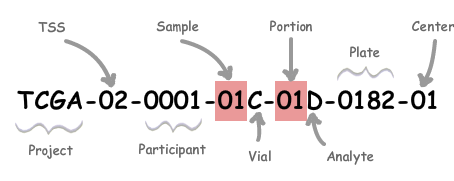
Moreover, I am currently analyzing the .txt data via excel, thus are there any other methods available instead? Any help will be greatly appreciated as I am currently quite lost, and worried about the lack of progress.
Thanks,
Ad.
