
I've made a simulation with a periodic boundary conditions - but lipid bilayer got assembled in such a way - that water (which is not shown in the picture below) stacked in the center of the box:
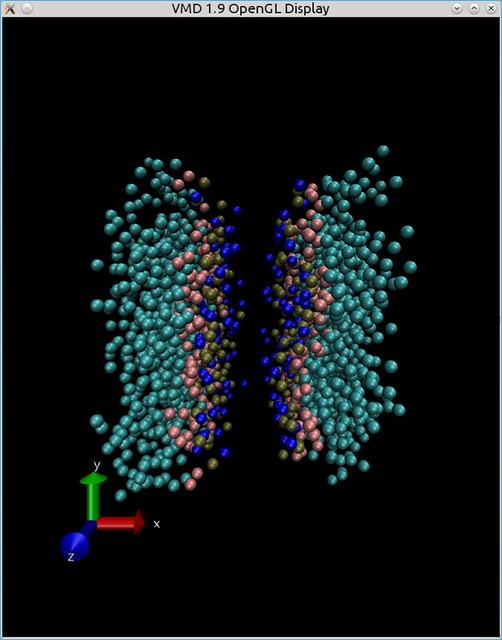
How do I move the box - so to make bilayer look more like a bilayer - in my cell-box?
Edit:
A warm thanks goes to comcon1.
In case one doesn't remember - one can find a box size by
gmxdump -f traj.trr | less
or
gmxdump -f traj.trr | vim -
to browse it in vim.
It said me that the initial box size was 7.5 7.5 7.5
So I've made a script to run through a range of x values (you must have confout.gro and topol.tpr files in the dir):
#!/usr/bin/python2.7
from numpy import arange
from os import system
for i in arange(-1.05, 5.05, 0.1):
system("editconf -f confout.gro -translate %g 0 0 -o edtOut%g.gro" %(i, i))
system("echo 0 | trjconv -f edtOut%g.gro -trans %g 0 0 -pbc res -o trjOut%g.gro" %(i, i, i))
This way I found that the bilayer is alright after the translations ranging from 0.05 to 3.25. So I've choosen 1.65 as a middle value. Than I've moved trajectory by:
echo 0 | trjconv -o trjOut1.65.trr -trans 1.65 0 0 -pbc res
To rotate a gro file - one should also first -rotate it with editconf, and then trjconv it with -pbc res:
editconf -f confout.gro -rotate 0 90 0 -o edtOut.gro
echo 0 | trjconv -f edtOut.gro -pbc res -o trjOut.gro
Of cource one can combine rotate and translate in the same shot.
It looks like there's no utility written to rotate trajectory trr file.

This is a very specialist question. Have you tried the Gromacs mailing list? - http://www.gromacs.org/Support
I went on to write a PyMOL script (here) - which translates the structure (either gro or pdb) to a given vector and generates the picture. Typical usage:
bk-pymol-pic.py -f confout.gro -s "resi 1-128" -x "-2" -y "-3" -z "0".