
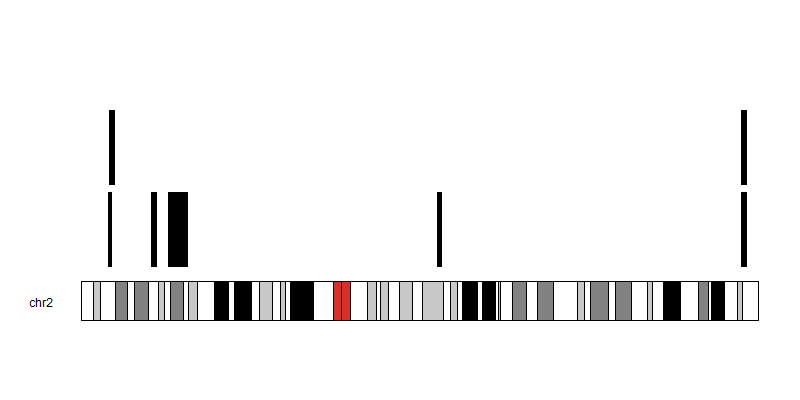

echo -e '@HD\tVN:1.4\tSO:unsorted\n@SQ\tSN:2\tLN:243199373' > tmp.dict && \
java -jar dist/biostar336589.jar -a 60 -R tmp.dict <(grep -v Chr input.bed) > out.svg
Entering edit mode

6.5 years ago
moithuti
•
0
I have genomic positions in a tab delimited file as follows:
Chr Start End
2 10262865 11801950
2 9637403 10927601
2 25141434 27157396
2 31181368 38044662
2 127808684 129276101
2 236957807 238896574
2 237172736 238896574
First I want to find overlapping segments within these genomic segments in the same file (edit). Then I wish to plot these such that the segments do not overlap on the plot. Is there a tool that can aid in:: 1 Finding overlapping regions within the same file 2. Visualising the segments in a non-overlapping manner?
My attempts at plotting this in R results in the segments occupying the same position
{kind=link}
What is the expected output plot for this data?
I would like to see each segment plotted without ovelarying each other, if the start positions for the segments are the same. Eventually I will then try to find the smallest region of overlap between the segments, so that if say I had 8 segments but the first 3 segments share an overlapping region I can find that region and have my segments reduced to 6 unique segments.
You should then try the
reduce()function fromGenomicRangesR package.I tried reduce() and it removes the metada making it hard for me to know which individuals share a given segment. Is there a way of retaining the metadata? From what I gather in the documentation, range reduction causes the names and metadata to be dropped. Or perhaps I should find a workaround for appending the metadata to the ranges?
you can start with that : https://stackoverflow.com/questions/20577492/finding-overlapping-regions-of-chromosome-with-granges