Entering edit mode

5.7 years ago
Assa Yeroslaviz
★
1.9k
We are running a ChIP-Seq experiment of multiple samples on one flow cell. when looking at the fastqc results of the samples, they all look strange.
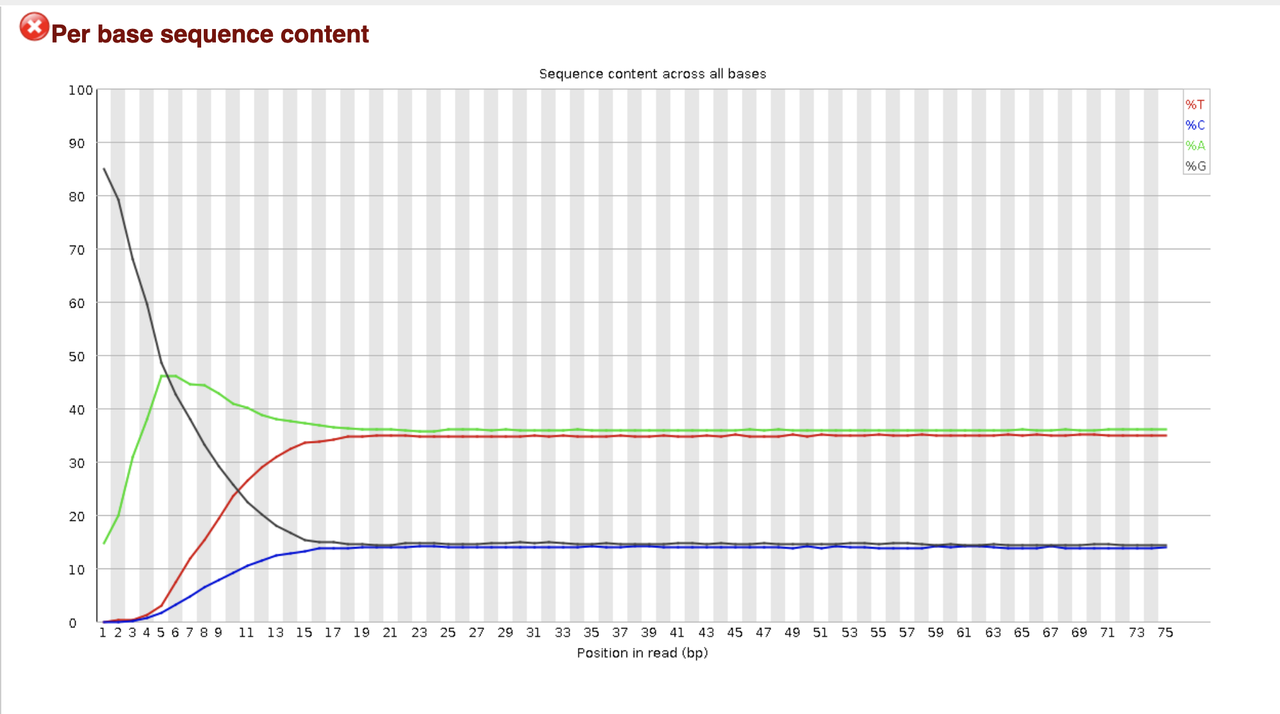
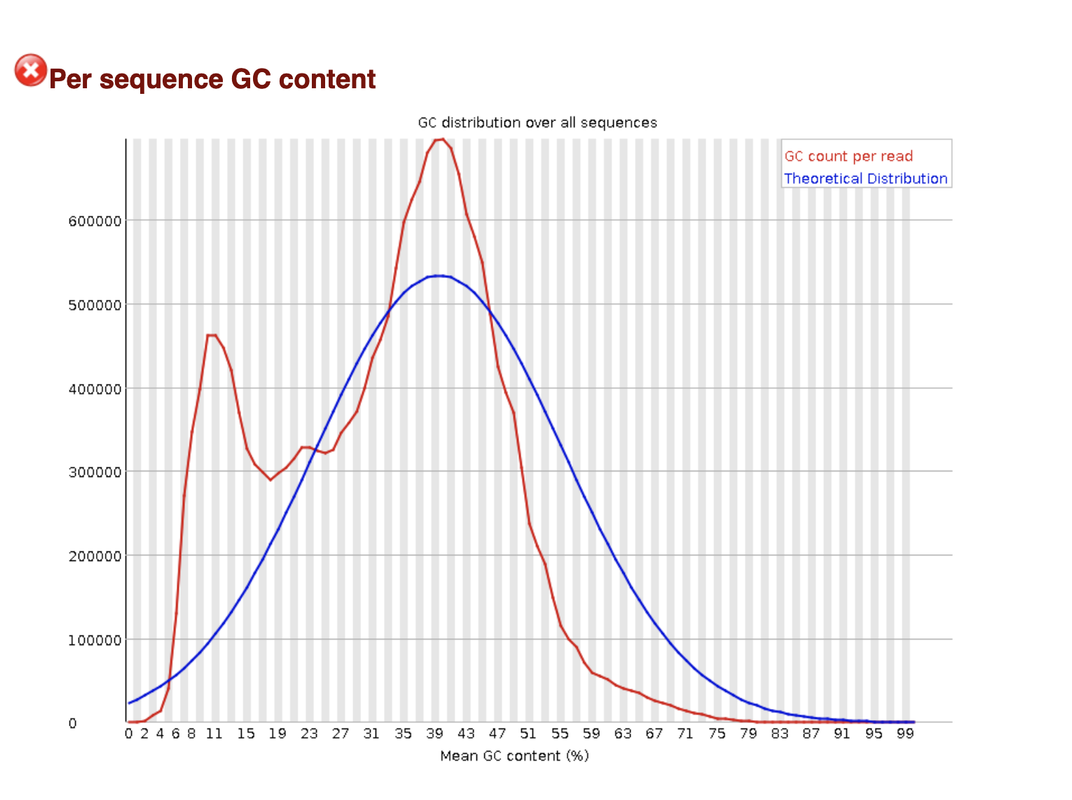
both overrepresented adapter and adapter content are in the green and sow no accumulation.
Can anyone explain this kind of behavior? Are these contaminations?
thanks
Have you scanned/trimmed this data? Is it from a 2-color chemistry sequencer (e.g. NovaSeq/NextSeq).
I haven't trimmed it yet. the sequencing was done a a nextseq with dual indexing.
About the trimming - do I just trim the first bases of the read (hard trim) or should I look for the barcodes (soft trim)?
Looks like you have a significant number of reads that may just be poly-G (certainly start with G, no signal = G) and those should be removed. Before you do any directed trimming why not just look for adapter sequence and see what happens. Any scan/trim program should work. Post plots after trimming.
This I have also considered, but why don't I see it also in the overrepresented sequences section? Is it possible that the adapter are so GC (or more G)-rich, that it skew the results so much?
Can we see what we get post-trimming? FastQC samples data for some of the features it looks at and does not include the entire dataset.