
Hi, friends I have finished call variants via GTAK best practices(joint-call) with 20 WES samples. My goal is to find the genes that may causes the disease from these 20 samples, I also tried some tools and methods, but I don't whether they are rigth. So, Is there a good guidance documentation like GTAK best practices to get my target?
I have tried an annotation tool named annova,
First, I transform vcf into the annova input format, more precisely, I got 20 annova input file for each sample.
convert2annovar.pl -format vcf4 relapse.filtered.snps.indels.vcf -allsample -filter PASS -out out/relapse
For each sample, I did the following:
1.filtering the irrelevant variants via 1000 Genomes Project dataset with MAF=0.01
2.annotating each variants with gene info, due to the data is WES, I got the "exonic variantfunction"
intronic KCNMA1 chr10 77008214 77008214 T C het 127.30 51
exonic CDHR4 chr3 49795287 49795287 C T het 8460.17 274
exonic CLCN5 chrX 50081733 50081733 A G hom 69769.27 255
intronic TUFT1 chr1 151566031 151566031 - C hom 7453.64 18
intronic PDE4D chr5 60147674 60147674 T G het 1524.08 112
intronic USF1 chr1 161041947 161041948 GA - het 2536.97 91
intronic RPS6KB2 chr11 67432552 67432552 A G het 2772.98 156
intergenic LINC01296(dist=73295),DUXAP10(dist=113998) chr14 19180792 19180792 C A het 2378.33 172
3.Combining there 20 output file with .MAF format to generate a waterfall plot, however, the percent of mutant is almost 100%, I don't think that is a right result.
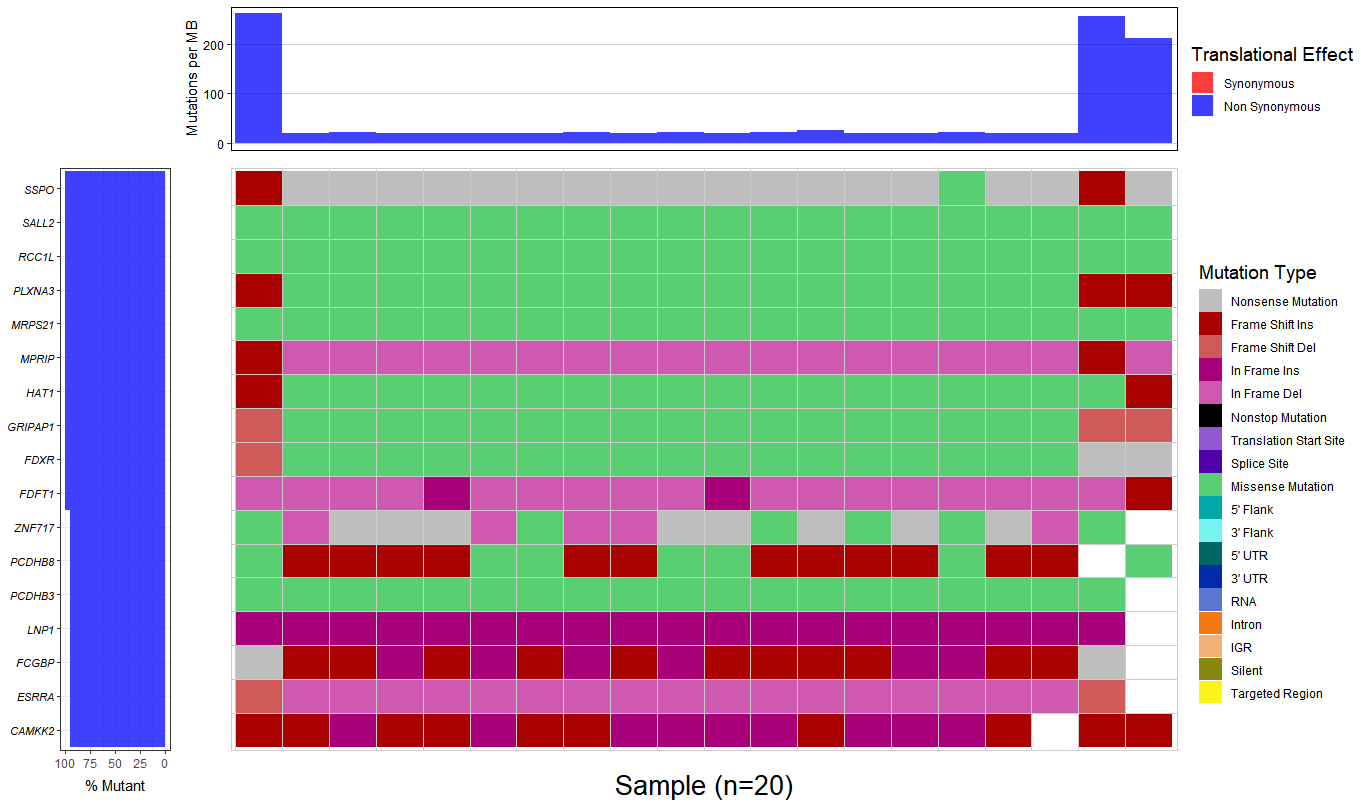

You should consider changing your question to the actual problem you are having, which is "find the genes that may causes the disease". GATK helps you get mutations, but it will not help you with understanding them.
Thanks for your reply, I added a description of the problem.
You mean GATK, right?
Yes, I got a vcf with gatk_v4.1.0.0, here is part of the output.
but I have no ideal what should I do to find these disease genes. is there a common practice ?
What have you tried?
Thanks for your reply, I added a description of the problem ,and I tried an annotation tool named 'annova'