Entering edit mode

5.4 years ago
Shicheng Guo
★
9.6k
Hi All,
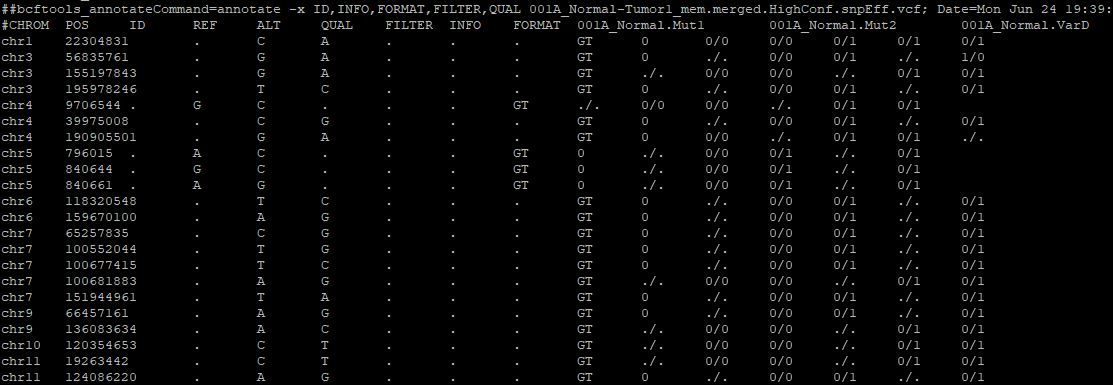
How to clean this strange VCF file? three different styles occurred: 0, 0/0 and ./.
Thanks.
Hi All,
How to clean this strange VCF file? three different styles occurred: 0, 0/0 and ./.
Thanks.
Use of this site constitutes acceptance of our User Agreement and Privacy Policy.
0/0 and ./. actually mean different things (this was a surprise to me at one point also). The ./. indicates no-call/no-call e.g. the variant caller made no determination, and 0/0 is homozygous ref. I don't know why you would have haploid things with just "0" in the first column
First, thanks so much for the help!
Second, I guess I can remove these columns with single '0', I am not sure how they come out. I guess maybe caused by windows/Mac/Linux file transfer? or stupid perl/python script converting?
Third, first three columns are mutation calling by 3 different method to same sample: Mut1, Mut2 and VarD. Here, I can merge these 3 columns to a consensus genotype. Do you have any idea to how to obtain the consensus genotype based on these 3 columns?
Thanks.
Shicheng
You will just have to create a rule and then filter based on that rule. For example, you can include a call if it is called in at least 1 sample, or specify that it must be called in all 3 [samples].
There are indirect ways of filtering for this via, for example, using bcftools to filter for the number of missing (
./.) and / or non-missing genotypes at each site.Define "clean" - what is the expected output?