I'm currently doing a differential expression analysis on a high number of samples (>100), mixing samples from different public datasets.
I've used Deseq2 so far, but I'm getting strange results, where some genes are given significantly differentially expressed, but with very high expression on few samples of one confition (around 5), and a low expression on all other samples. And I'm wondering if those results are just artifacts.
Would anybody know if Deseq2 is fitted to work with high number of samples? Or if another tool would be more relevant?
I am not so much concerned about the high number of samples, but more about how you combine different datasets. Tell us more about how you deal with the batch effects? Maybe a meta analysis would be more in place if the number of different studies used is also high.
For now I combined 3 studies, I've dealt with batch effect with the DEseq2 design (design = ~study + response), but I don't know to which extent it would correct the biases.
What do you mean by meta-analysis? Should I use specifics tools designed for meta-analysis?
Okay, if you are sure you can remove the batch effect properly you can continue with this approach. If you cannot you can do as ATpoint is suggesting (dataset-wise), or you can try meta-analysis (I am myself not a big fan of meta-analysis, but it is an alternative).
I am not a meta-analysis expert, but I have seen some RNAseq meta-analysis packages. What I understand from meta-analysis is that it is like your suggested approach (dataset-wise), followed with some additional statistics. Meta-analysis is often used for clinical studies.
Ok I get it! Out of curiosity, if there is no intersection in the results of all datasets analysed independently, is there any chance that I would find differentially expressed genes by merging all datasets and analyzing it as a whole?
Why can't this be biologically relevant? Many genes can be active or repressed under very limited conditions, see for example the famous Galactose expression system in yeast -> some random PNAS paper on Galactose induction in yeast (side note why would PNAS publish a Galactose induction study in 2015?)
Apology, I got carried away by the 2015 Galactose induction story - my intention was to make a similar point as Benn. The more studies you mix the more confounding factors you might have. You need to know the background of the studies to make sure your interpretation of the results is fair.
I agree with the two other comments, it is almost 100% certain (based on my experience) that you have notable batch effects induced by mixing different datasets that confound your results. I've seen strong batch effects with my own samples that I did myself here with the identical cell line but a few weeks apart, slightly different protocol and different sequencing platform. In PCA they cluster far apart and MA-plots look "all over the place" even though they should be pretty much identical. This effect is probably far more profound with datasets from different labs. I do not see how you could get meaningful results with this. Wouldn't it be better (if possible) to perform dataset-wise analysis and see if you get consistent results between the different analysis? That would save you from batch effects plus provide another level of confirmation.
Thank for your feedback. I indeed have batch effect, that I've tried to correct within my Deseq2 design (design = ~study + response).
When you mention data-wise analysis, do you mean that I should analyse each dataset separatly, and then cross the different results?
Exactly, each dataset independently. You can for a certain degree adjust for batch effects but I think adding study as a variable is by far not sufficient and you actually have no chance removing the batch effect. I would really do it as suggested. If results aggee between datasets it would be even more convincing to me as a reviewer as it ensures you are not chasing artifacts from one single study.
It is definitely good to have visualization before and after adjustment (even if that is really just median-centering independently calculated log2(FPKM + 0.1) expression before standardization in a heatmap, if you include batch in your differential expression model and then check if it worked).
If the batch effect is a huge problem (yet still randomized between studies), then the need to use a 2nd variable may be clear. Otherwise, you may want to check your consistency of replicates with 1-variable or 2-variables in the differential expression model.
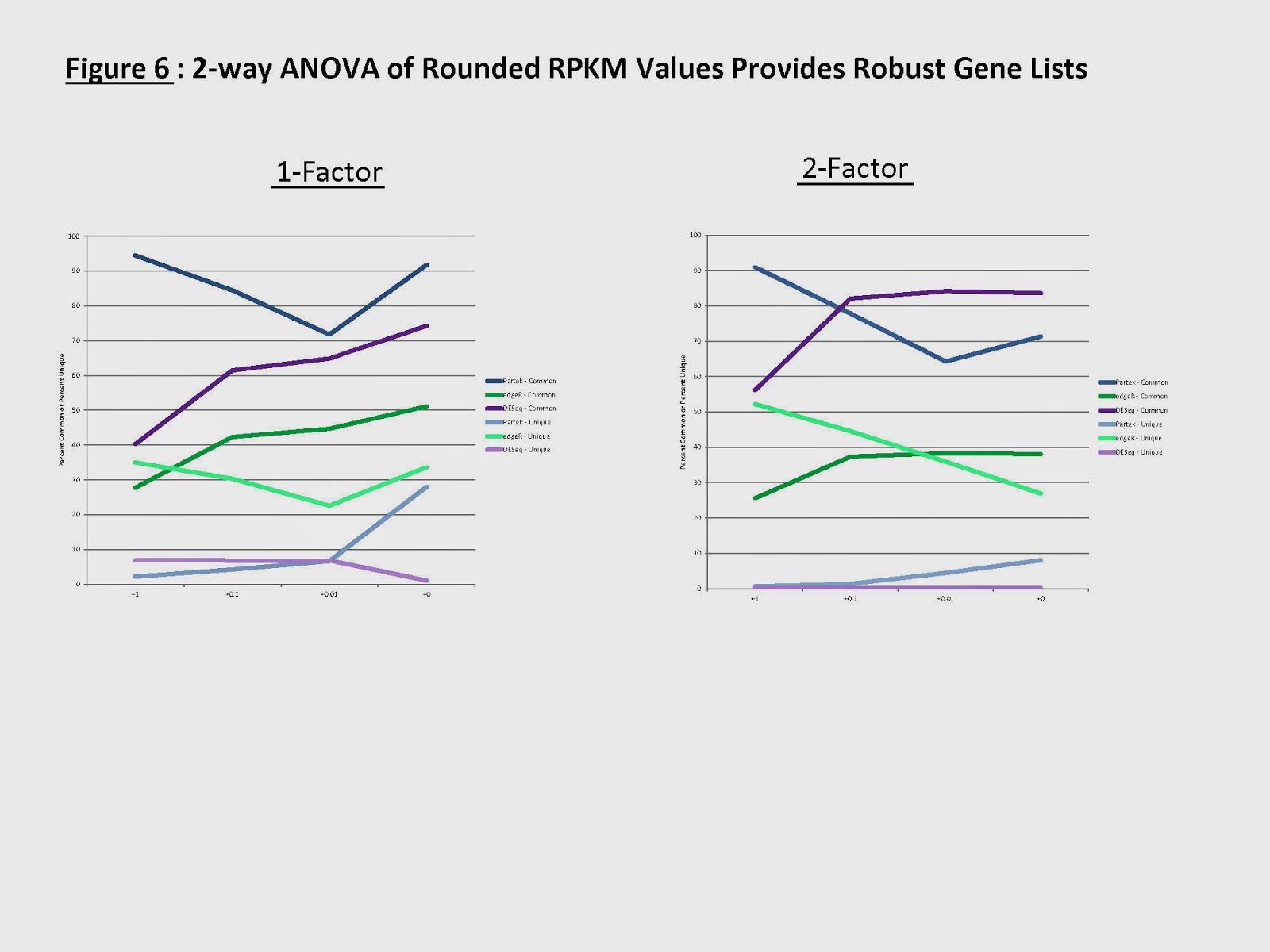
Even with large sample sizes, I know of at least one example where the genes that varied would still differ between programs (with >100 samples), and the relative ranking would even change depending upon whether a 1-variable or 2-variable model was used (Figure 6, which I copied over to a blog post, as the first image): http://cdwscience.blogspot.com/2013/11/rna-seq-differential-expression.html
[However, this was so long ago that I am using DESeq1 instead of DESeq2, even though DESeq1 may still be useful to have as an option for some projects]



I am not so much concerned about the high number of samples, but more about how you combine different datasets. Tell us more about how you deal with the batch effects? Maybe a meta analysis would be more in place if the number of different studies used is also high.
For now I combined 3 studies, I've dealt with batch effect with the DEseq2 design (design = ~study + response), but I don't know to which extent it would correct the biases. What do you mean by meta-analysis? Should I use specifics tools designed for meta-analysis?
Okay, if you are sure you can remove the batch effect properly you can continue with this approach. If you cannot you can do as ATpoint is suggesting (dataset-wise), or you can try meta-analysis (I am myself not a big fan of meta-analysis, but it is an alternative).
Out of interest, what do you mean by meta-analysis?
https://en.wikipedia.org/wiki/Meta-analysis
all hail wiki :)
Wiki oracle :)
I am not a meta-analysis expert, but I have seen some RNAseq meta-analysis packages. What I understand from meta-analysis is that it is like your suggested approach (dataset-wise), followed with some additional statistics. Meta-analysis is often used for clinical studies.
Ok I get it! Out of curiosity, if there is no intersection in the results of all datasets analysed independently, is there any chance that I would find differentially expressed genes by merging all datasets and analyzing it as a whole?
Merging datasets into one is only possible if you can correct for batch effect, so you'll need at least one overlapping group between all datasets.
Why can't this be biologically relevant? Many genes can be active or repressed under very limited conditions, see for example the famous Galactose expression system in yeast -> some random PNAS paper on Galactose induction in yeast (side note why would PNAS publish a Galactose induction study in 2015?)
Apology, I got carried away by the 2015 Galactose induction story - my intention was to make a similar point as Benn. The more studies you mix the more confounding factors you might have. You need to know the background of the studies to make sure your interpretation of the results is fair.