With regards to introns the answer depends on technology. For bulk-RNASeq data intronic reads are not counted. For single-cell RNASeq many tools count intronic reads as well (gives better results due to the low coverage in the first place).
With regards to reads falling outside of the CDS (aka in the UTR regions): Yes they are counted and should be as UTR regions are part of the mature mRNA.
In other words: the TPM / FPKM values measure exactly what you want :-)
Thank you for the answer. As a follow up, I am looking at RPF sequencing, specifically the EIF4E2 gene. Seems like a lot of reads map outside of CDS features in yellow. I hope these are not counted when it comes to expression of the gene itself. (read mapping and coverage )
I have to say I'm not an expert on ribo-seq so I don't know if that looks strange or not - also you don't show introns so it is a bit hard (but you could ask a new question specifically about that). In regards to whether they are counted or not it depends on the tool you are using - most bulk tools would not count introns but will count UTR regions (unless you tell them not to) - which tool are you using?
StringTie also counts UTR regions (but not introns (unless they are annotated as intron retentions)). If you only want to count CDS regions you can either use featureCounts or Kallisto/Salmon - but in both cases you would need to specify it's the CDS regions that should be quantified.


Thank you for the answer. As a follow up, I am looking at RPF sequencing, specifically the EIF4E2 gene. Seems like a lot of reads map outside of CDS features in yellow. I hope these are not counted when it comes to expression of the gene itself. (read mapping and coverage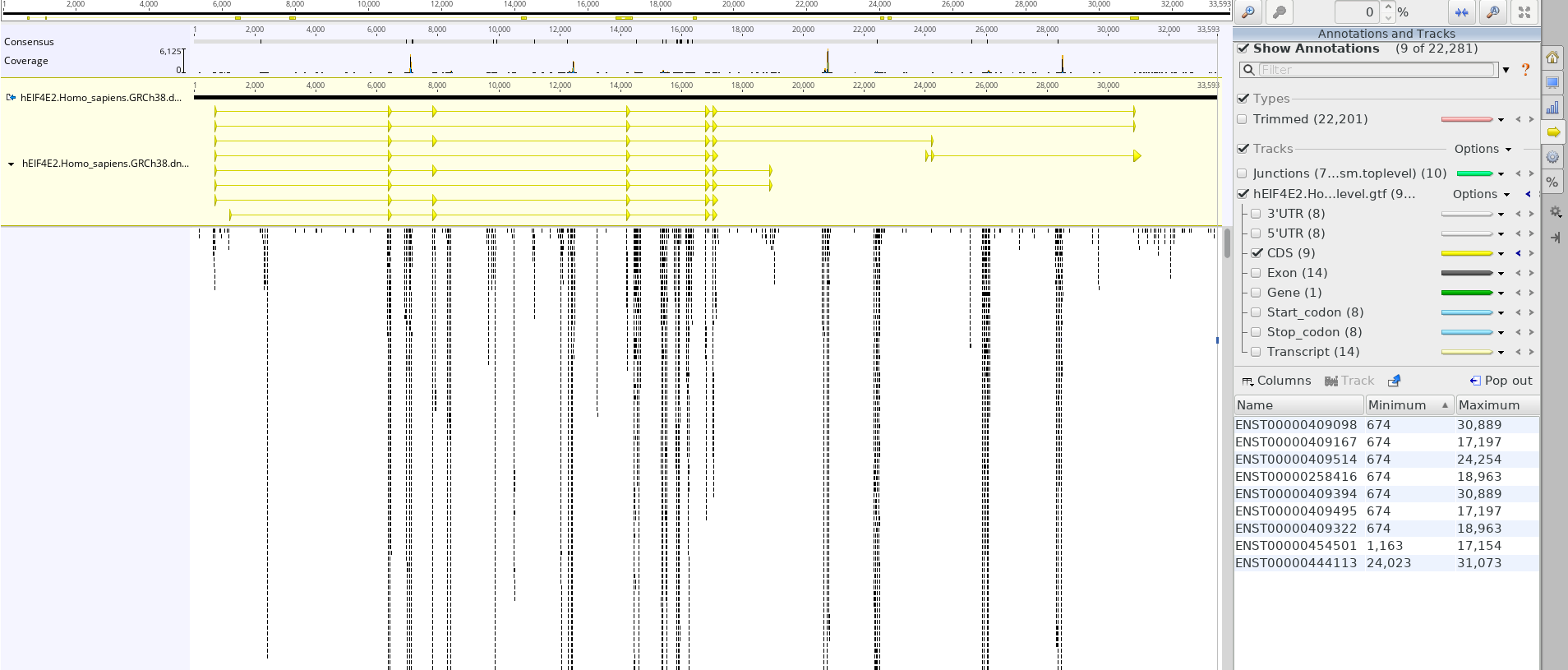 )
)
I have to say I'm not an expert on ribo-seq so I don't know if that looks strange or not - also you don't show introns so it is a bit hard (but you could ask a new question specifically about that). In regards to whether they are counted or not it depends on the tool you are using - most bulk tools would not count introns but will count UTR regions (unless you tell them not to) - which tool are you using?
I have to agree my title is a bit confusing. I have used HISAT to do the mapping but really the question is about stringtie that does the counting.
StringTie also counts UTR regions (but not introns (unless they are annotated as intron retentions)). If you only want to count CDS regions you can either use featureCounts or Kallisto/Salmon - but in both cases you would need to specify it's the CDS regions that should be quantified.