I am using TCGA ATAC data (bigwig files) to visualizing some peaks for some genes. In this paper there is a normalised count matrix file for each peak and sample? In supplementary file they mentioned that “To get the number of Tn5 insertions per peak, each corrected insertion site (end of a fragment) was counted…”. https://science.sciencemag.org/content/sci/suppl/2018/10/24/362.6413.eaav1898.DC1/aav1898_Corces_SM.pdf
So my question is that what is this count matrix and Tn5 insertions? Is there any relationship between peak height and number of Tn5 insertions? Can I use this number (Tn5 insertions) to select significant peaks ?
I do not know how much biological background you have so I answer a bit more extensively:
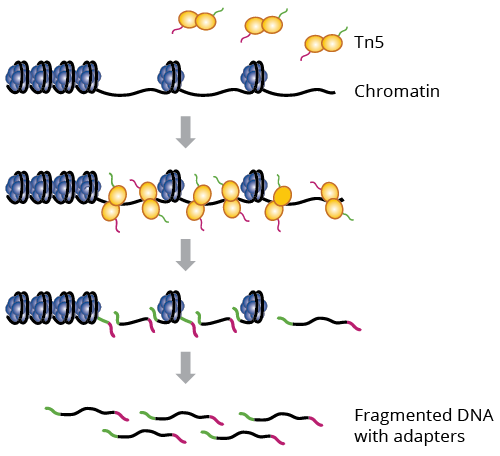
The Tn5 is the workhorse of ATAC-seq. This enzyme is added to the native chromatin and will insert an Illumina adapter to the DNA at sites that are not protected by nucleosomes while simultaneously fragmentating the DNA. DNA in open chromatin is therefore adapter-tagged and can be enriched over the background of closed chromatin using PCR followed by quantification via NGS.
The accumulation of Tn5 insertion sites is therefore a measure of chromatin accessability. From the technical site, given you have the Tn5 insertion positions (=the 5' end of each read) you can use standard peak callers to identify local enrichments (=peaks). One typically extends the Tn5 sites by like 50bp in each direction to smoothen the signal and allow more precise identification of peak summits.
The count matrix is then simply created by intersecting peak locations and Tn5 sites (or reads which is basically the same).
So yes peak height is a function of Tn5 insertion frequency (which is the same as read counts).
So number in count matrix is "number of Tn5 insertions" , If this number is high then peak height also would be high and this is probability for open chromatin structure. Am I right ? Can I use this numbers to find differential significant peaks between two class of samples?


Nice explanation thank you so much,
So number in count matrix is "number of Tn5 insertions" , If this number is high then peak height also would be high and this is probability for open chromatin structure. Am I right ? Can I use this numbers to find differential significant peaks between two class of samples?
Again thanks a lot
Yes one could use the count matrix but one would probably use raw counts to feed it into tools like
edgeR. See if they provide raw counts.