Entering edit mode

5.1 years ago
poltora4enko
▴
10
Hello!
I'm trying to use bowtie2 with options bowtie2 -p 8 -a -x bt2_file -U file.fq -S file.sam, because I need multiple alignment for my chip-seq, but it is very slowly. How can I make it faster? I already tried options --very-fast , because quality is not so important for me, anyway it's very slowly. May be I need to use some more options or another tools ? Thanks for attentions! and sorry for bad English!
Alignment is limited by disk and CPU speed. If this is what your machine can do at best, then this is it, not much you can do about it. If you have the resources you can parallelize several jobs with GNU parallel. Beyond that, get coffee and wait or simply run overnight.
Thank you for answer! I already thought that I was doing something wrong
Bowtie2 is kind of old now and definitely not the fastest tool out there. If you want to stick with traditional alignment, HISAT2 is a good replacement (from the same group) and is already an order of magnitude faster. See figure 1 of this paper.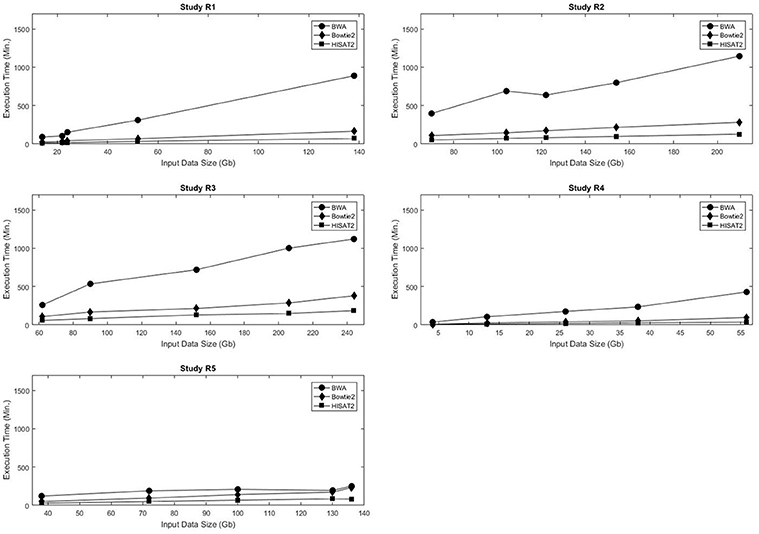
hmm,clearly . I thought about it, it's worth a try, thanks!!!!!!!