Entering edit mode

5.0 years ago
marongiu.luigi
▴
730
Hello,
I have generated synthetic reads with ART running the following command:
# concatenate individual reference fasta for each chromosome
# here mutations were introduced
# and I made one fasta for each allele, thus there are two independent files for each chromosome
cat chr01a.fa ... > chrA.fa
cat chr01b.fa ... > chrB.fa
# generate fastq
# ~/src/art/Illumina_profiles/custom/HiSeq2k_0m1.txt gives the profile for the output.
art_illumina -1 ~/src/art/Illumina_profiles/custom/HiSeq2k_0m1.txt -2 ~/src/art/Illumina_profiles/custom/HiSeq2k_0m2.txt \
-p -f 30 -l 140 -m 300 -s 10 -i chrA.fa -o chrA_
art_illumina -1 ~/src/art/Illumina_profiles/custom/HiSeq2k_0m1.txt -2 ~/src/art/Illumina_profiles/custom/HiSeq2k_0m2.txt \
-p -f 30 -l 140 -m 300 -s 10 -i chrB.fa -o chrB_
# concatenate
cat chrA_1.fq chrB_1.fq > chr_1.fq
cat chrA_1.fq chrB_1.fq > chr_2.fq
gzip chr_1.fq
gzip chr_2.fq
# align
bwa mem -t 12 GRCh38.fa chr_1.fq.gz chr_2.fq.gz | samtools view -Sb - | samtools sort -o AlnSrt.bam
sambamba markdup -r -t 12 --overflow-list-size 1000000 --hash-table-size 1000000 --tmpdir=./TEMP AlnSrt.bam AlnSrtDed.bam
I then checked the alignment with Qualimap but I got this figure:
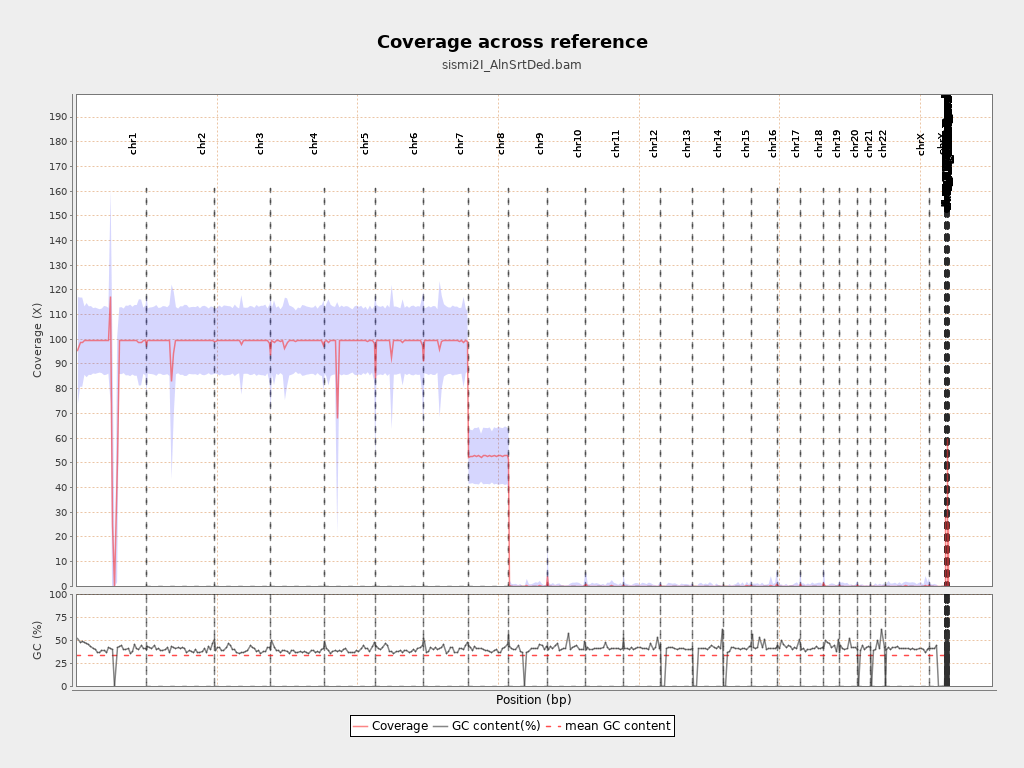 Is it normal that there is a drop in coverage after chromosome 7?
Is it normal that there is a drop in coverage after chromosome 7?
Is it normal the negative spike in coverage in chromosome 1?
I even did the art routine one chromosome at the time and then merged the fastq files but I got:
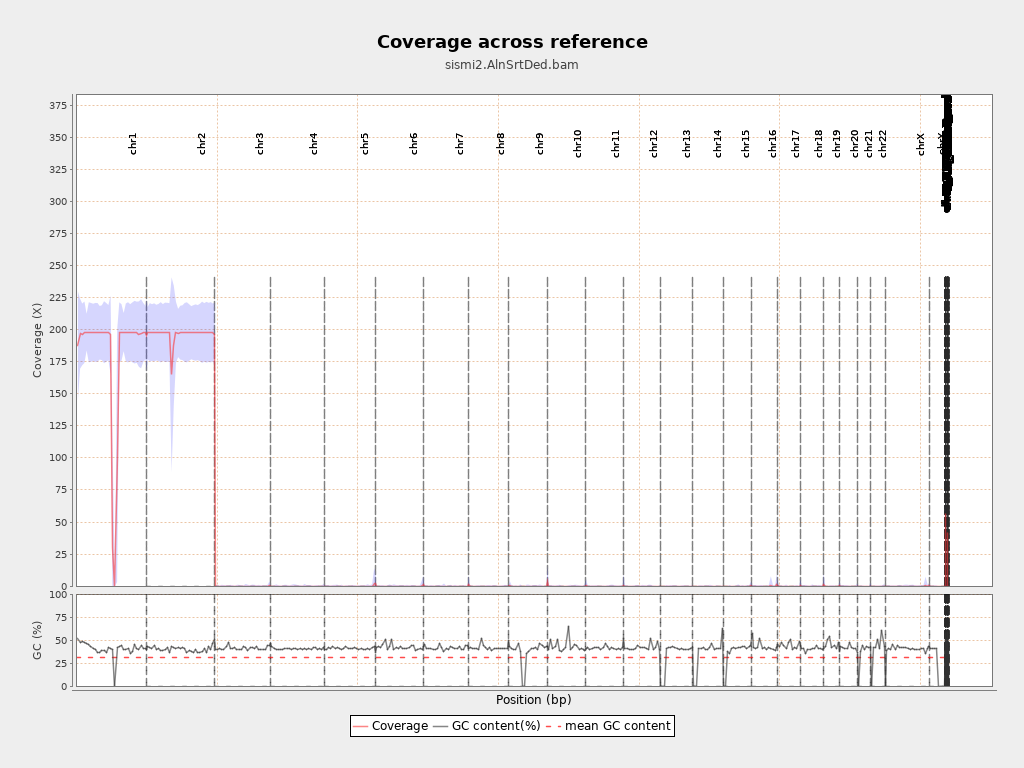 which is even worse.
which is even worse.
Thanks
Have you tried to generate the reads from the entire genome at one time. Like you would in a real experiment. One does not isolate chromosomes and sequence them independently.
Negative spike at 1: centromere with 0 coverage (it is long there)
Positive spike chr1 left to centromere: polymorphic CNP region
Drop exactly 0.5x at chr8: aneuploidy (doubt that it is survivable) or you just forget to switch in one of the files (more likely)
Ok that makes sense, but what about the other (9-23)? did I forget to put on all of them?
yesss. Or at some point your aligner e.g. gave up since it does not have enough memory (happens with me a lot when I use SLURM). you see coverage around 0 - these are 0-mapping-quality reads and it is perfectly normal to have them.
It is likely that @manongiu has run these jobs independently since working with one chromosome at a time?
I did not get error during the alignment, but let's say I will use a cluster for the alignment instead of my commodity PC: then the drop should disappear? (having the cure to double check the presence of all chromosomes, of course).
Or is it better to run the alignment one chromosome at the time and then merge the bam files with
samtools merge merged.bam in.1.bam in.2.bam ...? Would that cause problems with the chromosome indexing or something? Tx