Entering edit mode

4.9 years ago
Shicheng Guo
★
9.6k
Hi All,
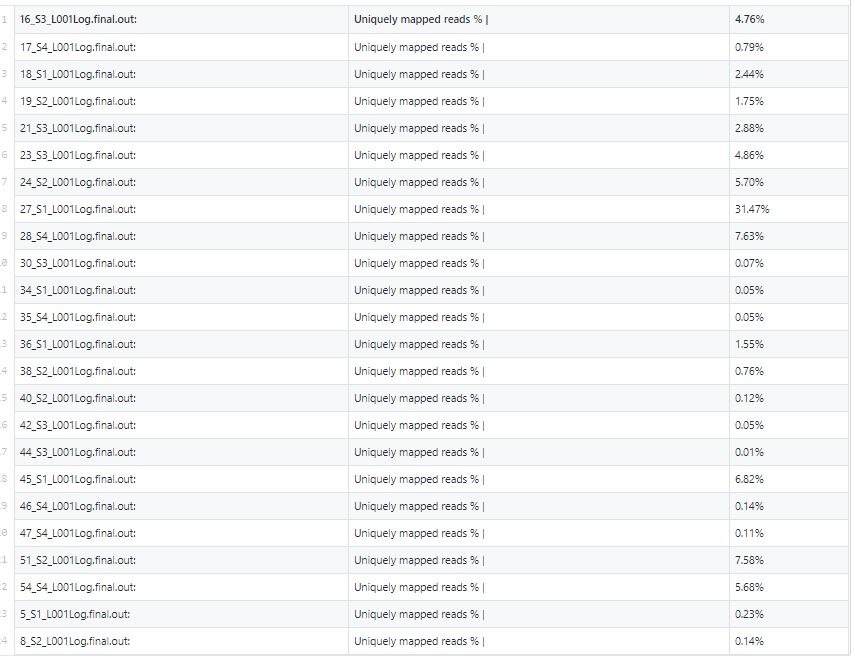
I apply STAR (latest version: 2019) to map human RNA-seq data and I found the uniquely mapping ratio is highly variable ranging from 0.01% to 30%. Any suggestions or comments on such a weird high variable mapping ratio?
Thanks
Solved: the samples are special (only small part is human RNA).
STAR --runThreadN 24 --outSAMtype BAM SortedByCoordinate
--outBAMsortingThreadN 6 --genomeDir $DBDIR --outFileNamePrefix ~/hpc/project/RnaseqBacterial/extdata/rnaseq/$i --readFilesIn $i\_R1_001.fastq $i\_R2_001.fastq