Entering edit mode

4.8 years ago
Evan
▴
250
Hello, I'm new on single-cell analysis and the use of deconvolution methods.
I would like to create my own signature matrix from single-cell rna data to use it in Cibersortx as a reference profile. Currently, I'm using Seurat to cluster my cells in cell type following this tutorial : https://satijalab.org/seurat/v3.1/pbmc3k_tutorial.html
Is it possible to get a table with in column the cells labeled with their cell type and in rows the genes with their expression in each cells (Count/RPKM/TPM ?).
In fact I would like a table which look like the picture below to use it as single cell reference sample file to build a signature matrix file to use in Cibersortx.
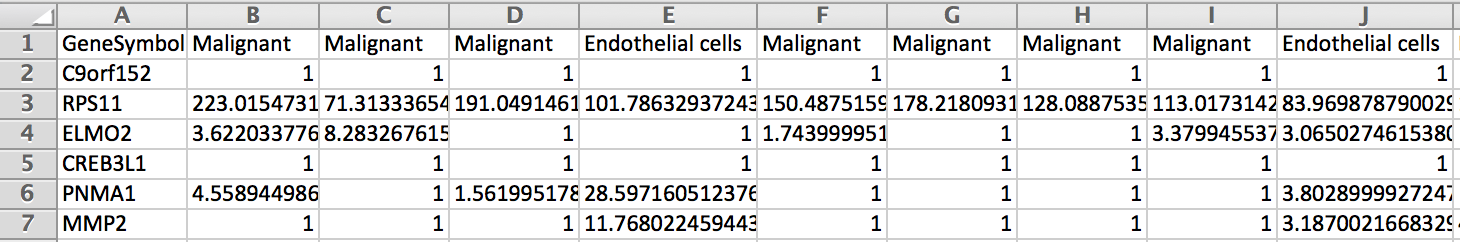
I would be very grateful if someone could explain me how to do it. Thank you.


Thank you for sharing. I face the same problem. Can you share Python script which can merge these two files? Thanks a lot!
Hi, I also have the same problem. Did you find a solution?
Hi, sorry for the delay. Please find the code in my answer for oomoru.
Hi Evan, as you exported the raw counts (stored in
pbmc@assays[["RNA"]]@counts), did you normalize your reference sample file prior creation of the signature matrix (e.g., RPKM) or did you submit the raw counts? Did you make any filtration on raw data prior creation of the signature matrix? ThanksHi, at the moment I don't normalize my Raw Counts matrix, as it's mentionned on Cibersortx tutorial (on it's website), raw counts are recommended. Don't forget that to get the bests results as possible, your signature matrix and your Bulk RNA-Seq must be in the same space normalization (in my case I use Raw Counts). I will not recommand RPKM to perform cellular deconvolution even if Cibersortx is able to convert it into TPM.
Read this excellent article to know more about that :
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7648640/
> Did you make any filtration on raw data prior creation of the signature matrix?
No more than the filtrations I made in Seurat to process the data.
Hello, please can you share the Python script for merging the files? Thanks.
Hi, here is the code, it's way too long for a simple thing but it has the advantage to be fast and the ram consumption is very low compared to pandas or R.
Thank you very much, this is so helpful to me. I used to the code but had error with this section-
KeyError Traceback (most recent call last)
<ipython-input-105-3265a1ddf5eb> in <module> 11 split_first_line_cell_sequence = first_line_cell_sequence.rstrip("\n").split("\t") 12 for element in split_first_line_cell_sequence: ---> 13 header.append(dict_sequence_label[element]) 14 file_matrix.write("GENES\t"+'\t'.join(header)+"\n") 15 # Once I wrote all the cells label I can write the gene counts
KeyError: '1_AAACCTGAGACTTGAA.1'
I don't know why it keeps giving the error. Please, how can I avoid this. I appreciate your time and kind assistance.
Hi, it seems the cells abels in the file "Convert_UMI_Label.tsv" are not the same than in the raw counts matrix "Gene_Count_Per_Cell.tsv". Maybe, in one of these file the cell label (barcode) is 1_AAACCTGAGACTTGAA.1 and in the other "1_AAACCTGAGACTTGAA_1". Could you give me one cell barcode per file to adjust the code for you ?
Thanks Evan! Gene_Count_Per_Cell.tsv, first barcode- 1_AAACCTGAGACTTGAA.1 Convert_UMI_Label.tsv, first barcode- 1_AAACCTGAGACTTGAA.1
The barcodes are the same for both files. At first, I was getting an error that stated- KeyError: ' ', when my files looked like this-
so I adjusted them in excel to this-
Then the error became this- KeyError: '1_AAACCTGAGACTTGAA.1' - this is the first barcode in both files.
I think the error is occurring because we are taking elements from the dictionary which has the quotation mark as headers in the new file, but I don't know much, and how to go about it.
Hum okay I see, could you upload the both files somewhere to help you ?
Thanks a lot Evan! I have the files in the link below. https://drive.google.com/drive/folders/1GkwubWLgyUgStveemX43Ud4l1_e5Hnt9?usp=sharing
-it is publicly available data from geo ncbi.
Thanks ! I modified some stuff in your files, firstly, I removed the " in the Convert_UMI_Label.tsv, secondly the 'X' in front of the barcodes in the Gene_Count_Per_Cell.tsv and finally the the tabulation before the first barcode in the count matrix.
I ran the script after these little modifications and the (pre) signature matrix has been generated without problems :)
Here is the link where you can download the corrected files and the (pre)signature matrix generated : https://mega.nz/folder/REUBzCaZ#uhFr6F82imiky2LVVf1nSg
Now you're able to build your signature matrix with Cibersortx. You can do it on their website (storage limited to 1Gb and this sigmatrix is ~ 670mb) or directly from Docker. You must request a token before using Docker. Enjoy !
Thank you very much Evan!