
Hi everyone,
I am trying to found out what residues are in contact with a ligand (the binding site) in a PDB file. To give you an example, I need to find the residues in contact with FMN ligand in the PDB entry 1AL7.
First of all, can we find this type of information in a PDB file? Or do we need a software to calculate it? I think the first question is yes, but I am not sure: I used the new mmCIF format, as they are well organized compared to the old version. In this format, there is a Data Category called "chem_comp" and the dictionnary on the PDB website says :
"Data items in the CHEM_COMP category give details about each of the chemical components from which the relevant chemical structures can be constructed, such as name, mass or charge." It allows me to know if there is the presence of a FMN ligand.
Another category is "struct_site":
"Data items in the STRUCT_SITE category record details about portions of the structure that contribute to structurally relevant sites (e.g. active sites, substrate-binding subsites, metal-coordination sites)."
In my example, there is a line "AC1 Software ? ? ? ? 22 'BINDING SITE FOR RESIDUE FMN A 360'"
And then, you have another category called "struct_site_gen":
"Data items in the STRUCT_SITE_GEN category record details about the generation of portions of the structure that contribute to structurally relevant sites."
This category is, I guess, my answer, because I see all the residues with their number sharing the same id (AC1) than in struct_site.
Can I use this information for the binding site or you guys think I have to use a software to calculate the residues in the binding site? If so, which one?
Thank you


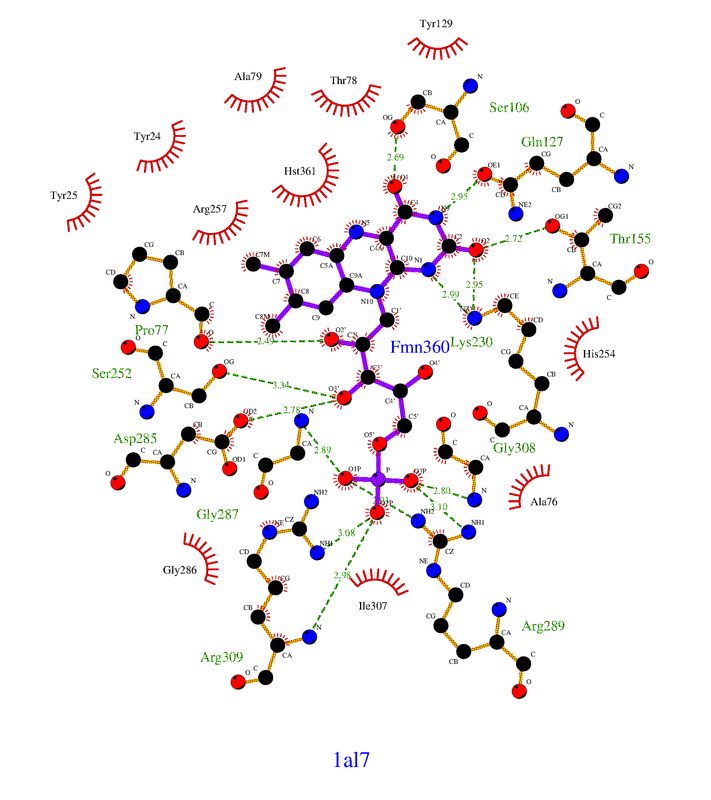
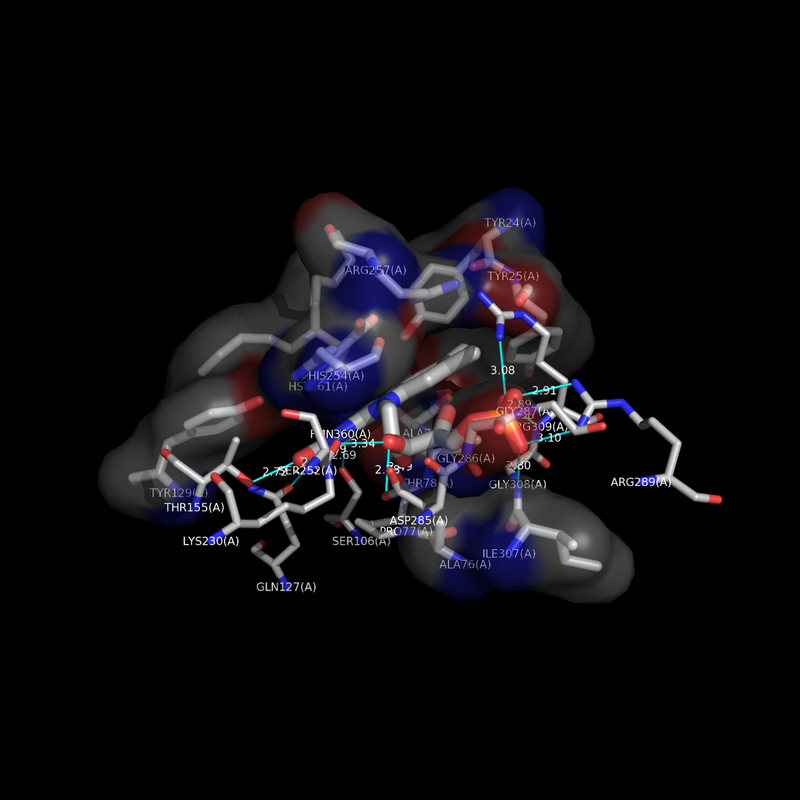
Hello, I was wondering if you had success in finding the non-bonded contact residues programatically? I am performing similar research and would greatly appreciate if you could share any success you have had regarding this task!